

Summary
The primary mechanisms supporting the immunoregulatory polarization of myeloid cells upon infiltration into tumors remain largely unexplored. Elucidation of these signals could enable better strategies to restore protective anti-tumor immunity. Here, we investigated the role of the intrinsic activation of the PKR-like endoplasmic-reticulum (ER) kinase (PERK) in the immunoinhibitory actions of tumor-associated Myeloid-derived suppressor cells (MDSC). PERK signaling increased in tumor-MDSC, and its deletion, transformed MDSC into myeloid cells that activated anti-tumor CD8+ T-cell immunity. Tumor-MDSC lacking PERK exhibited disrupted NRF2-driven anti-oxidant capacity and impaired mitochondrial respiratory homeostasis. Moreover, reduced-NRF2 signaling in PERK-deficient MDSC elicited cytosolic mitochondrial-DNA elevation, and consequently, STING-dependent expression of anti-tumor Type-I Interferon. Re-activation of NRF2 signaling, conditional deletion of STING, or blockade of Type-I interferon receptor-I restored immunoinhibitory potential of PERK-ablated MDSC. Our findings demonstrate the pivotal role of PERK in tumor-MDSC functionality and unveil strategies to reprogram immunosuppressive myelopoiesis in tumors to boost cancer immunotherapy.
Keywords: Tumor immunity, MDSC, PERK, Unfolded protein responses, NRF2, STING, Type I IFN
Graphical Abstract

eTOC/“In Brief”:
The mechanisms underlying the augmented immunoinhibitory activity of Myeloid-derived suppressor cells (MDSC) upon infiltration into tumors remain elusive. Mohamed and colleagues demonstrate that the unfolded protein responses (UPR)-related kinase, PERK, promotes tumor-MDSC functionality through stimulation of the transcriptional factor NRF2, which thereby restricts the immunostimulatory axis, cytosolic Mitochondrial-DNA: STING: Type-I-IFN.
Introduction
The myelopoiesis process that protects against tumors is drastically derailed in most individuals with advanced malignancies towards the expansion of myeloid subsets that block protective anti-tumor T cell immunity and promote cancer cell progression (Singhal et al., 2016). Expansion of myeloid-derived suppressor cells (MDSC), a heterogeneous group of monocytic (M-MDSC) and polymorphonuclear (PMN-MDSC) precursors, has emerged as a key mechanism of anti-tumor immune evasion (Veglia et al., 2018) and correlates with poor clinical outcome and resistance to cancer immunotherapy (Lu et al., 2017; Weber et al., 2016). Upon infiltration into the tumor microenvironment (TME), MDSC amplify their immunoregulatory potential through upregulation of Arginase I and Nitric oxide synthase 2 (NOS2), release of reactive oxygen species (ROS) and peroxynitrite, and production of several regulatory cytokines (Gabrilovich et al., 2012). Despite their relevance in tumors, there are limited strategies to inhibit the detrimental activity of MDSC in part because of the incomplete understanding of the central processes governing the MDSC functionality within the TME.
Multiple cytokines, growth factors, and stress-related conditions in the TME polarize tumor-MDSC into highly immunosuppressive cells (Mohamed et al., 2018). However, the mechanisms by which MDSC evolve to augment their immunosuppressive potential in the harsh tumor milieu remain elusive. Activation of the unfolded protein responses (UPR) controls the quality of the proteome pool in cells experiencing endoplasmic reticulum (ER) stress as the result of the accumulation of misfolded proteins in the ER (Bettigole and Glimcher, 2015). Priming of the UPR also functions as a physiological signaling program in cells undergoing high secretory and metabolic activity triggered by cytokines, Toll-like receptors agonists, or other stimuli (Janssens et al., 2014). The UPR is characterized by the orchestrated stimulation of the activating transcription factor-6, the inositol-requiring enzyme 1 (IRE1α), and the protein kinase RNA (PKR)-like ER kinase (PERK, encoded by Eif2ak3) (Walter and Ron, 2011). Activation of PERK occurs through oligomerization and auto-phosphorylation, leading to the phosphorylation of multiple targets, including the transcription factor NF-E2-related factor 2 (NRF2, encoded by Nfe2l2) (Pytel et al., 2016). In tumor cells, PERK-linked phosphorylation of NRF2 induces cellular redox transcripts that relieve the effects of ROS (Cullinan et al., 2003). Although stimulation of the UPR modulates the survival of cancer cells in the TME (Chevet et al., 2015), their role in the immunoinhibitory activity of myeloid cells in tumors is incompletely understood. Seminal reports have highlighted the impact of IRE1α, XBP1 and the unresolved ER stress sensor, C/EBP homologous protein (CHOP, encoded by Ddit3) in the immunoregulatory activity of myeloid cells in tumors (Condamine et al., 2016; Cubillos-Ruiz et al., 2015; Thevenot et al., 2014; Yan et al., 2016). However, the mechanisms by which the activation of the UPR, and in particular PERK, controls the immunoregulatory activity of MDSC in tumors remain unknown.
The stimulator of interferon genes (STING, encoded by Tmem173) represents a major mediator of innate immunity triggered by DNA (Ishikawa and Barber, 2008). Primed STING recruits and promotes the phosphorylation of the TANK-binding kinase 1 (TBK1), which phosphorylates the interferon regulatory factor 3 (IRF3), thereby promoting the production of Type I interferon (IFN) (Ahn et al., 2014). Although STING activation in myeloid cells has emerged as a key driver of anti-tumor immunity (Woo et al., 2014), the pathways regulating the intrinsic activation of STING signaling in tumor-MDSC continue to be poorly understood.
Here, we sought to elucidate the role of PERK in the immunosuppressive activity of tumor-MDSC. Deletion of PERK re-routed tumor-MDSC into cells that primed anti-tumor CD8+ T cell immunity by reducing NRF2 signaling, which thereby triggered STING-dependent production of Type I IFN through accumulation of cytosolic mitochondrial DNA (mtDNA). Our results suggest the targeting of PERK as a strategy to overcome immunosuppressive myelopoiesis in tumors and to enhance the effects of cancer immunotherapy.
Results
TME promotes immunoregulatory activity and UPR activation in MDSC
To elucidate the impact of the UPR priming in the regulatory activity of MDSC in tumors, we first monitored the crosstalk between the T cell inhibitory potential and the activation of UPR sensors in CD11b+Gr1+ cells from tumors (T-MDSC) and spleens (S-MDSC) of mice bearing s.c. Lewis lung carcinoma (LLC), s.c. B16 melanoma, or orthotopic ID8-Defb29-Vegfa ovarian tumors; and in splenic myeloid counterparts (iMC) from tumor-free mice. A superior capacity to impair T cell proliferation and an increased expression of total and phosphorylated PERK and IRE1a were detected in tumor-MDSC compared to splenic-MDSC or iMC (Figure 1A–B), without a distinct accumulation of MDSC subsets (Figure S1A). Also, consistent with a preferential ER enlargement in tumor-MDSC, elevated fluorescence of ER tracker (Hambrock et al., 2002) (Figure 1C, Figure S1B–C), and augmented ER dilation detected by transmission electron microscopy (Figure 1D) were found in tumor-MDSC compared to splenic-MDSC or iMC. Next, we tested whether the exposure of bone marrow-derived MDSC (BM-MDSC) to tumor explants (TES) triggered activation of the UPR and immunoregulatory potential. Pre-treatment of murine BM-MDSC with LLC-TES (Thevenot et al., 2014), or human BM-MDSC with supernatants from the renal cell carcinoma cell line 786–0 (hTES) (Rodriguez et al., 2009), resulted in upregulation of total and phosphorylated PERK and IRE1a and enhanced ability to block T cell proliferation, which were similar to the effects induced by the UPR inducer, Thapsigargin (Thaps) (Figure 1E–H). Furthermore, administration of Thaps into LLC-bearing mice promoted tumor growth, splenic-MDSC expansion, and tumor-MDSC immunosuppressive function (Figure S1D–F). These results suggest the potential link between UPR activation and immunoinhibitory activity in tumor-MDSC.
Figure 1. TME drives immunosuppressive function and UPR activation in MDSC.
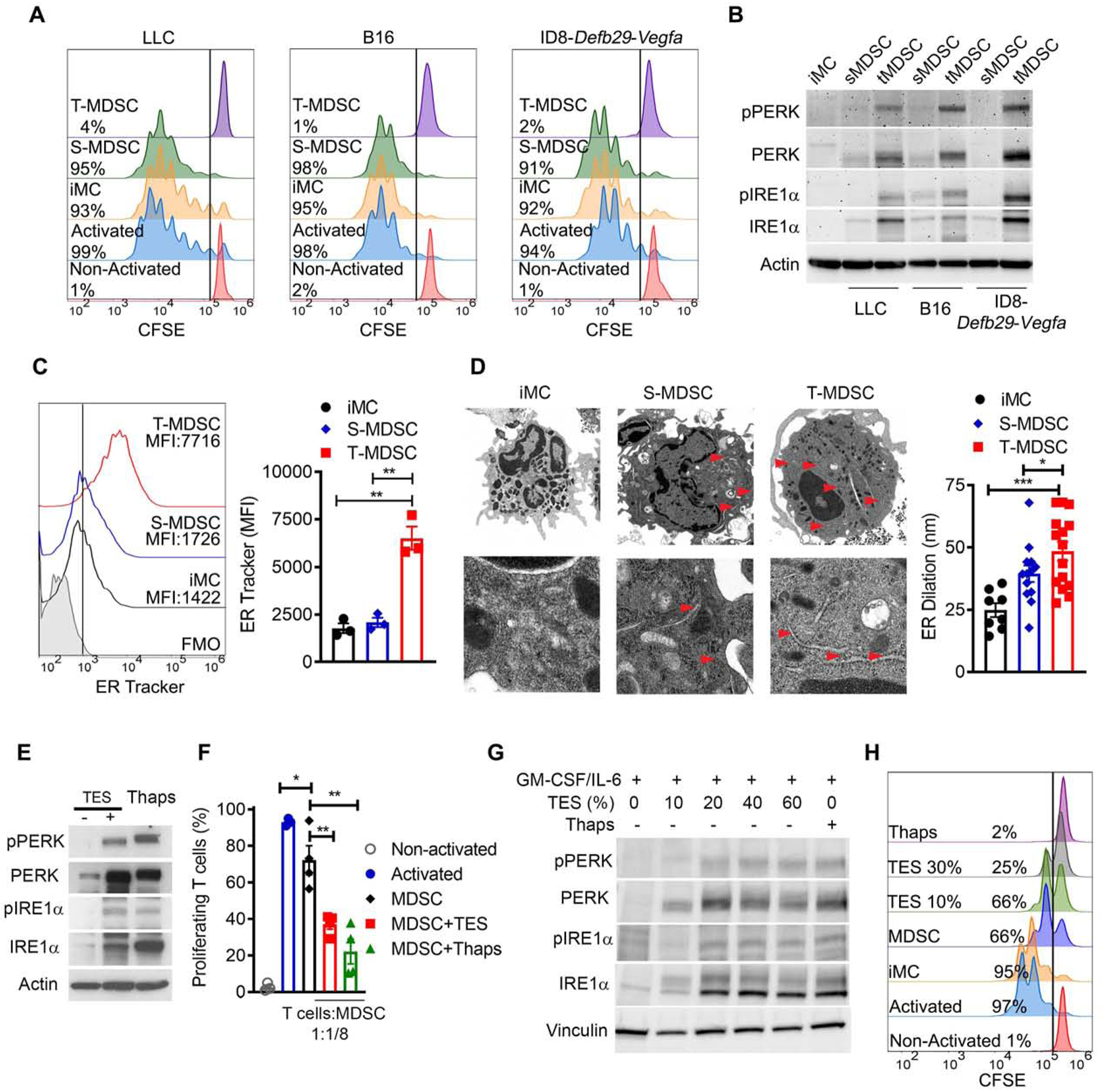
(A) Proliferation assessed by flow cytometry in CFSE-labeled T cells primed with anti-CD3 and anti-CD28 and co-cultured for 72 hours with S-MDSC or T-MDSC from mice bearing LLC (left), B16 (middle) or ID8-Defb29-Vegfa (right) tumors; or from splenic iMC from tumor-free mice (1:1/4 ratio). Sample from 3 independent repeats.
(B) Immunoblot for the UPR sensors in iMC, S-MDSC, and T-MDSC from (A). Results are representative of 3 independent studies.
(C) Sample of ER-Tracker compared to fluorescence minus one (FMO) from 3 distinct repeats (left) and merged mean fluorescence intensity data (MFI, right) in iMC, S-MDSC, or T-MDSC from LLC-bearing mice.
(D) Transmission electron microscopy image from 3 independent repeats from cells as in (C) (left). Arrows indicate ER. Scale bar, 2 μm (upper) and 200 nm (lower). Quantification of data (right).
(E-F) BM-MDSC derived using G-CSF and GM-CSF (20 ng/ml each) and in the presence of LLC-TES (30%) or Thaps (200 nM, 24 hours) were tested for the UPR drivers (E) or capacity block T cell proliferation (ratio 1:1/8) (F). Immunoblot and suppression assay are representative of 3 distinct repeats.
(G-H) Human-MDSC derived using GM-CSF and IL-6 (10 ng/ml each) and in the presence of 10–60% supernatants from the 786–0 cell line or Thaps were tested for the expression of UPR drivers (G) or ability to blunt primed T cell proliferation (ratio 2:1) (H). Immunoblot and histograms are representative from 5 independent repeats.
Statistics were done using one-way ANOVA or Student’s t-test. *, p<0.05; **, p<0.01. Please also see Figure S1.
Mitigation of UPR blunts MDSC activity and boosts cancer immunotherapy
We determined whether the inhibition of the UPR activation impacted tumor growth and MDSC-linked T cell dysfunction using Tauroursodeoxycholic acid (TUDCA), a molecular chaperone that alleviates ER stress and UPR priming (Nakagawa et al., 2014; Ozcan et al., 2006). Treatment of mice bearing established LLC or B16 tumors with TUDCA delayed tumor growth (Figure 2A), which correlated with a decreased ability of tumor-MDSC to block T cell proliferation (Figure 2B, Figure S2A), and reduced expression of the MDSC-inhibitory factor Arginase I, but not NOS2 (Figure 2C). Also, in support of the mitigation of UPR by TUDCA, lower expression of phospho-PERK and phospho-IRE1a was noticed in tumor-MDSC from TUDCA-treated mice, compared to MDSC from controls (Figure S2B). Because the activity of MDSC depends on tumor burden, we then evaluated the effect of TUDCA in cultured MDSC. TUDCA reduced the ability of human BM-MDSC conditioned with hTES, murine BM-MDSC developed after exposing BM-iMC to TES, and LLC tumor-MDSC to block T cell proliferation (Figure S2C–E). Next, we sought to establish the role of T cells in the anti-tumor effects induced by TUDCA. Higher expansion of recently activated CD69+CD44+CD8+ T cells was detected in tumors from B16 or LLC-bearing mice treated with TUDCA (Figure 2D). Moreover, the anti-tumor actions triggered by TUDCA were not observed in immunodeficient Rag1−/− mice (Figure 2E), suggesting the key role of T cells in the anti-tumor effects induced by TUDCA.
Figure 2. TUDCA overcomes MDSC-related T cell dysfunction and boosts immunotherapy.
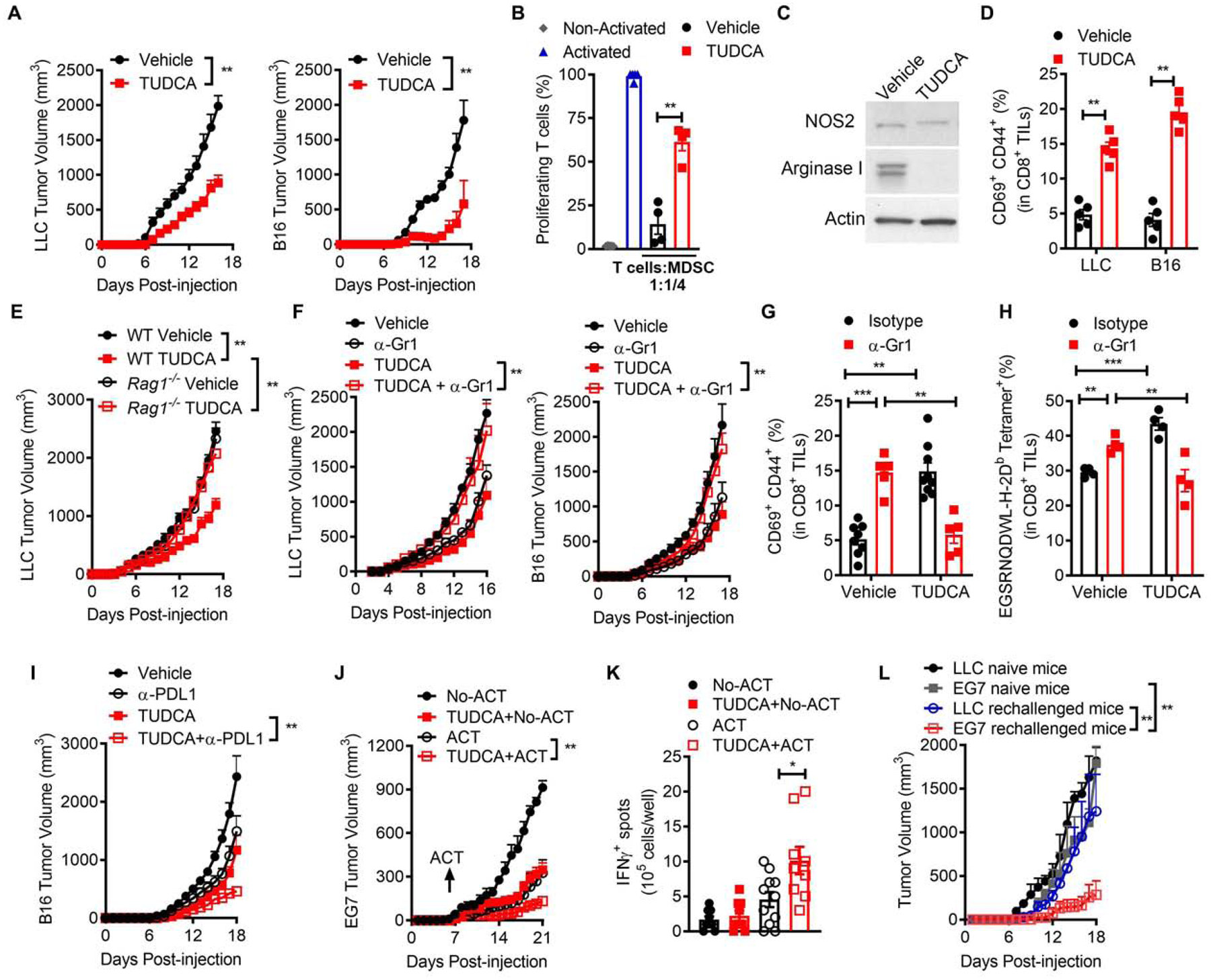
(A) Tumor volume ± SEM in mice bearing LLC (left) or B16 (right) cells and treated daily after day 6 post-tumor injection with vehicle or TUDCA (250 mg/kg). n=10.
(B) Proliferation of primed CFSE-labelled T cells co-cultured with tumor-MDSC (ratio 1:1/4) from LLC-bearing mice treated with vehicle or TUDCA. n=5.
(C) Immunoblot for NOS2 and Arginase I in tumor-MDSC from (A). n=3 independent repeats.
(D) Percentage of CD69+CD44+ in CD8+ TILs from tumors as in (A) at day 15. n=5.
(E) LLC tumor volume ± SEM in wildtype (WT) and Rag1−/− mice treated as in (A). n=5.
(F) Tumor volume ± SEM in mice bearing LLC (left) or B16 (right) tumors treated as in (A) and receiving or not 250 μg anti-Gr1 every 3rd day since the day of tumor injection. n=5.
(G-H) Percentage of CD69+CD44+ (G) and EGSRNQDWL-H-2Db tetramer+ (H) in CD8+ TILs from B16-bearing mice treated as in (F).
(I) B16 tumor volume ± SEM in mice treated as in (A) and receiving or not 250 μg anti-PD-L1. n=10.
(J) Mice bearing EG7 tumors were treated as in (A) and specific cohorts received CD8+ OT-I T cells pre-primed with OVA257–264. Tumor volume ± SEM. n=5.
(K) Spleens from (J) tested for IFNγ by EliSpot upon priming with OVA257–264. n=3 repeats of 3 spleens/group.
(L) Tumor volume ± SEM for EG7 or LLC tumors injected s.c. in opposite flanks of mice that previously rejected EG7 tumors after TUDCA plus ACT treatment. n=3.
Statistics were applied using one-way ANOVA or student’s t-test, *, p<0.05; **, p<0.01. Please also see Figure S2.
Next, we studied whether treatment of tumor-bearing mice with TUDCA switched MDSC into cells that elicited anti-tumor responses. Depletion of MDSC-like cells upon injections with anti-Gr1 antibody delayed B16 and LLC tumor progression in vehicle-treated mice, while restoring tumor growth in TUDCA-treated counterparts (Figure 2F). Moreover, anti-Gr1 or TUDCA treatments promoted the spontaneous expansion of recently primed CD69+CD44+CD8+ T cells and EGSRNQDWL-H-2Db-tetramer+CD8+ T cells recognizing the gp100 melanoma antigen in B16-bearing mice (Figure 2G–H). Conversely, the increase of these tumor-reactive T cell groups was prevented in TUDCA-treated mice after co-administration with anti-Gr1 (Figure 2G–H), showing that TUDCA treatment transforms tumor-MDSC into T cell-stimulating cells.
Furthermore, we tested whether TUDCA amplified the effects of various forms of immunotherapy. Treatment with TUDCA plus anti-PD-L1 induced synergistic anti-tumor effects in B16-bearing mice, compared to mice treated with the single agents (Figure 2I). Also, we evaluated the effect of TUDCA in an adoptive T cell transfer (ACT) model against the experimental antigen ovalbumin (OVA), in which pre-activated anti-OVA257–264 (SIINFEKL) OT-I cells were transferred into mice bearing OVA+ EG7 tumors. Higher anti-tumor responses were noted in mice treated with TUDCA plus ACT, compared to those receiving TUDCA or ACT single treatments or untreated controls (Figure 2J), which correlated with a higher frequency of IFNg-producing T cells upon activation of splenocytes with SIINFEKL (Figure 2K). Moreover, some of the mice receiving TUDCA plus ACT rejected the EG7 tumors and were resistant to re-challenge with the same tumor cells, but not against non-related tumors (Figure 2L), indicative of specific anti-tumor protective memory. Thus, our results suggest that TUDCA enhances the effects of immunotherapy in tumor-bearing mice.
PERK regulates the immunosuppressive activity of tumor-MDSC
To monitor the UPR activation status in human MDSC infiltrating tumors, we tested the expression of phospho-PERK in tumor-linked MDSC from a TMA created from 46 patients with metastatic non-small cell lung carcinoma (Met-NSCLC) through automated multispectral imaging. Higher phospho-PERK was detected in cells resembling PMN-MDSC (Pan-CytokeratinnegCD11b+HLA-DRnegCD14negCD15+) and M-MDSC (Pan-CytokeratinnegCD11b+HLA-DRnegCD14+CD15neg) from Met-NSCLC tumors, compared to counterparts from control tissues (Figure 3A). In addition, we validated the activation of PERK in MDSC from a TMA from 84 patients with high-grade serous ovarian cancer. Elevated frequency of phospho-PERK+ M-MDSC-like cells was found in ovarian tumors, compared to same subsets in healthy ovaries (Figure S3A). However, we could not consistently analyze phospho-PERK expression in PMN-MDSC from ovarian tumors as limited infiltration of cells resembling PMN-MDSC was noted (Figure S3A). Together, our results indicate the elevated PERK activation in tumor-MDSC from human malignancies.
Figure 3. PERK intrinsically controls MDSC function.
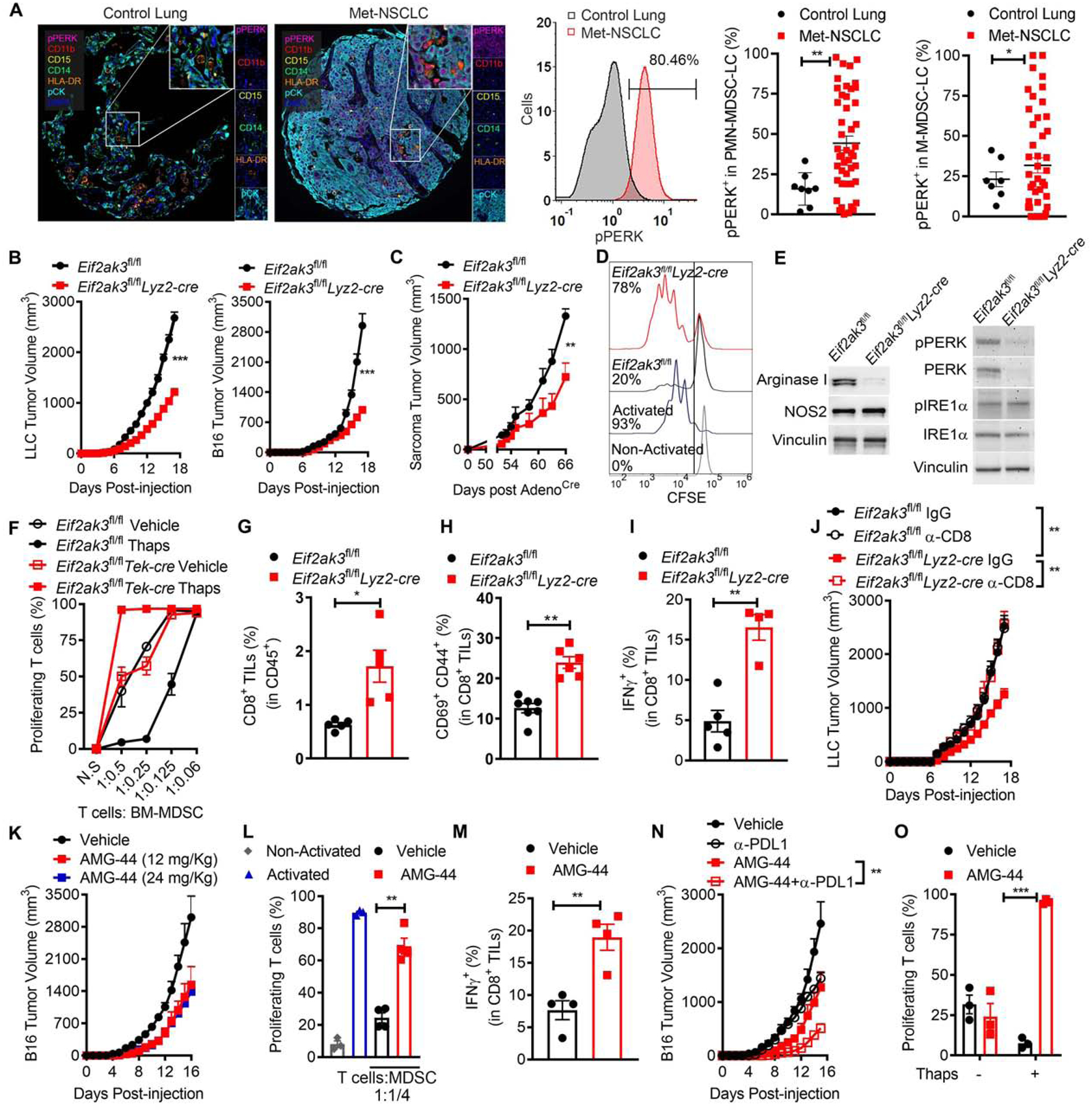
(A) Left: Illustrative image of 46 Met-NSCLC tumors and 8 healthy control lung tissues from a TMA (20x resolution and further 10x digital magnification) showing phospho-PERK (Magenta), CD11b (Red), CD15 (Yellow), CD14 (Green), HLA-DR (Orange), pan-Cytokeratin (pCK, Cyan), and DAPI (Blue) by Automated Multispectral Imaging. Center: Sample histogram from samples from left showing pPERK expression on pCKnegCD11b+HLA-DRneg cells from MET-NSCLC and control lungs. Right: Phospho-PERK+ cells ± SEM in PMN-MDSC-LC (pCKnegCD11b+HLA-DRnegCD14negCD15+) or M-MDSC-LC (pCKnegCD11b+HLA-DRnegCD14+CD15neg). n=8 control lung tissues and 46 MET-NSCLC tumors.
(B) LLC (left, n=20) and B16 (right, n=10) tumor growth ± SEM in Eif2ak3fl/fland Eif2ak3fl/flLyz2-cre mice.
(C) Growth of flank sarcoma initiated after injection with Cre recombinase-coding adenovirus in flank of KrasG12D/+Trp53fl/fl mice previously reconstituted with bone marrow from Eif2ak3fl/flLyz2-cre or Eif2ak3fl/fl mice. n=4.
(D) Proliferation of primed CFSE-labeled T cells co-cultured with tumor-MDSC (ratio 1:1/4) from LLC-bearing control or PERK-null mice. Results are representative from 3 independent experiments.
(E) Immunoblots for NOS2 and Arginase I (left) and UPR mediators in LLC tumor-MDSC from (D). Representative from 3 distinct experiments.
(F) Control and Eif2ak3−/−BM-MDSC pre-treated or not with Thaps (24 hours) were co-cultured with primed-T cells. T cell proliferation was assessed as in (D). n=4.
(G-I) Percentage of CD8+ TILs in CD45+ cells (G); and CD69+CD44+ (H) and IFNγ+ cells (I) in CD8+ TILs in LLC tumors as in (B). n=5.
(J) LLC growth ± SEM in Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice treated with 400 μg anti-CD8 antibody or isotype. n=5 mice/group.
(K) Tumor volume ± SEM in B16-bearing mice treated with vehicle or AMG-44 (12, 24 mg/kg) daily after day 6 post-tumor injection. n=5.
(L) Proliferation of primed T cells co-cultured with tumor-MDSC (ratio 1:1/4) from B16-bearing mice treated with vehicle or AMG-44 (12 mg/kg). n=3.
(M) Frequency of IFNγ+ in CD8+ TILs from B16-bearing mice treated as in (L). n=4.
(N) B16 growth ± SEM in mice treated as in (L) and with or without 250 μg anti-PD-L1. n=5.
(O) Proliferation of primed T cells co-cultured with control or Thaps-treated BM-MDSC (ratio 1:1/8) pre-exposed to 5 μM AMG-44. n=3.
Statistics were by one-way ANOVA, Student’s t-test, or log-rank (Mantel-Cox)*, p<0.05; **, p<0.01. Please also see Figure S3.
To study the impact of PERK in MDSC on tumor growth, we generated myeloid cell-conditional Eif2ak3−/− mice, after breeding Eif2ak3fl/fl mice with mice expressing lysozyme (Lyz2)-driven Cre recombinase (Eif2ak3fl/flLyz2-cre), which enabled gene excision in tumor-associated M-MDSC, PMN-MDSC, and macrophages, and to a lower extent in myeloid DCs (Figure S3B–C). Delayed growth of LLC and B16 tumors and prolonged survival after injection with ID8-Defb29-Vegfa ovarian tumors were observed in Eif2ak3fl/flLyz2-cre mice compared to controls (Figure 3B, S3D). To expand our data in a spontaneous tumor model, lethally-irradiated LSL-KrasG12D/+Trp53fl/fl mice received bone marrow cells from Eif2ak3fl/flLyz2-cre or Eif2ak3fl/fl mice. Seven weeks later, mice were injected with intra-muscular adenovirus coding for Cre-recombinase and followed for growth of autochthonous flank sarcomas, which depend on MDSC activity (Rutkowski et al., 2015). Delayed tumor progression was found in LSL-KrasG12D/+Trp53−/− mice reconstituted with bone marrows from Eif2ak3fl/flLyz2-cre mice, compared controls (Figure 3C). Also, a reduction in the ability to blunt T cell proliferation and in the expression of PERK and Arginase I, but not in IRE1α or NOS2, were detected in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice, compared to MDSC from Eif2ak3fl/fl mice (Figure 3D–E). Moreover, higher expansion of cells resembling M-MDSC, but not PMN-MDSC, was observed within tumors from Eif2ak3fl/flLyz2-cre mice (Figure S3E), which did not correlate with changes in the apoptosis marker Annexin V, or the unmitigated ER stress driver of apoptosis, DR5 (Lu et al., 2014) (Figure S3F–G). To rule out the effect of the tumor burden differences in the actions induced by PERK deletion, we obtained BM-MDSC from hematopoietic-endothelial specific PERK-null mice (Eif2ak3fl/flTek-cre), as Eif2ak3 excision was inefficient in BM-MDSC from Eif2ak3fl/flLyz2-cre mice (Figure S3H–K). Supporting the intrinsic role of PERK in UPR-primed MDSC, excision of Eif2ak3 abolished the amplified T cell-inhibitory effect found in BM-MDSC treated with Thaps, without altering the regulatory potential of vehicle-treated BM-MDSC (Figure 3F). Furthermore, elimination of PERK did not impact the frequency of common myeloid progenitors (CMP) or granulocyte-macrophage progenitors (GMP) (Trento et al., 2017) in the bone marrow of tumor-bearing mice nor affected MDSC development from hematopoietic precursors (Figure S3L–M). Because of the role of MDSC in T cell dysfunction, we then studied the effect of T cells in the anti-tumor responses observed in Eif2ak3−/− mice. Higher frequency of total, recently activated, and IFNg-expressing CD8+ T cells was found in tumors from Eif2ak3fl/flLyz2-cre mice, compared to controls (Figure 3G–I). Also, depletion of CD8+ T cells restored tumor growth in Eif2ak3fl/flLyz2-cre mice (Figure 3J), indicating the protective CD8+ T cell immunity status in tumor-bearing mice lacking PERK in myeloid cells.
To recapitulate our findings in therapeutic models, we next evaluated the anti-tumor effects of two small-molecule PERK inhibitors, GSK-2606414, a dual PERK and RIPK1 inhibitor (Rojas-Rivera et al., 2017), and AMG-44, a potent and selective PERK inhibitor (Smith et al., 2015). Delayed tumor growth was noticed in B16-bearing mice treated with GSK-2606414, which correlated with a lower immunoinhibitory activity of MDSC (Figure S3N–O). A primary limitation for the use of GSK-2606414 is the induction of pancreatic toxicity (Yu et al., 2015). Therefore, we tested the therapeutic action of two different doses of AMG-44. A similar delay in tumor growth, without alterations in the blood glucose concentration or in the size or insulin expression in the pancreatic islets were found in B16-bearing mice treated with both AMG-44 doses (Figure 3K, Figure S3P–R). Also, illustrating the therapeutic potential of AMG-44, treatment of B16-bearing mice with AMG-44 impaired the immunoregulatory activity of tumor-MDSC, induced expansion of tumor-infiltrating CD8+ T cells (TILs) expressing IFNγ and synergized with anti-PD-L1 therapy (Figure 3L–N). Moreover, the anti-tumor action of AMG-44 was detected in PERK-competent mice, but not in Eif2ak3fl/flLyz2-cre counterparts (Figure S3S), suggesting the role of myeloid cell-PERK in the effects induced by AMG-44. Next, we tested the intrinsic effect of AMG-44 on Thaps-treated MDSC. Similar to Eif2ak3−/− MDSC (Figure 3F), AMG-44 overcame the ability of Thaps-treated BM-MDSC to impair T cell proliferation, without affecting MDSC baseline activity (Figure 3O). Overall, these results demonstrate the key role of PERK in MDSC activity and the therapeutic potential of PERK inhibitors to overcome MDSC-linked T cell dysfunction.
PERK deletion reprograms tumor-MDSC into immune-stimulatory cells.
We evaluated whether the elimination of PERK transformed MDSC into cells that promoted anti-tumor T cell responses. Deletion of MDSC-like cells after continuous injections with anti-Gr1 or anti-CCL2 antibodies restored LLC tumor growth in Eif2ak3fl/flLyz2-cre mice, while delaying tumor growth in Eif2ak3fl/fl mice (Figure 4A–B). Also, co-injection of tumor-infiltrating Eif2ak3−/− CD11b+ Gr1+ cells plus LLC cells into C57BL/6 mice (WT), but not into Rag1−/− mice, resulted in slower tumor growth, compared to LLC cells implanted alone or mixed with control tumor-MDSC (Figure 4C). Next, we compared the expression of major inflammatory drivers in tumor M-MDSC and PMN-MDSC from LLC-bearing Eif2ak3fl/flLyz2-cre and Eif2ak3fl/fl mice. Higher expression of MHC-I (H-2Kb), CD40, and CD80; but not MHC-II, CD11c, and CD86 was found in PERK-null M-MDSC compared to controls, whereas no changes were noted on PMN-MDSC (Figure 4D). Moreover, elevated expression of anti-tumor cytokines, interleukin-12 (IL-12) and TNFα, was detected in tumor M-MDSC and PMN-MDSC from Eif2ak3fl/flLyz2-cre mice (Figure 4E). Next, we tested whether the deletion of PERK allowed tumor-MDSC to process and present exogenous antigens. PERK-deficient tumor-MDSC pulsed with complete OVA showed higher ability to prime OT-I T cell proliferation, though lower than DCs, compared to control MDSC (Figure 4F). To further study the ability of Eif2ak3−/− MDSC to engulf tumor products and to cross-present antigens in the TME, we used injections with Pan02-OVA-ZsGreen tumors, engineered to express fluorescence protein ZsGreen and OVA. Elevated ZsGreen and SIINFEKL-bound H-2Kb were detected on tumor-MDSC from Eif2ak3fl/flLyz2-cre mice compared to controls (Figure 4G–H, Figure S4A), which correlated with an extension of OVA-reacting SIINFEKL-H-2Kb-tetramer+ CD8+ TILs (Figure 4I). PERK deletion promoted the expression of ZsGreen and SIINFEKL-bound H-2Kb in tumor M-MDSC and PMN-MDSC, but not in macrophages or myeloid DCs, indicating a preferential effect on MDSC subsets (Figure 4J–K). Also, in agreement with the ability of PERK-ablated MDSC to present engulfed tumor antigens to CD8+ T cells, tumor-CD11b+Gr1+ cells from Eif2ak3fl/flLyz2-cre mice bearing Pan02-OVA-ZsGreen tumors, but not from control mice, induced ex vivo proliferation of OT-I CD8+ T cells (Figure 4L), but not of OT-II CD4+ T cells (Figure S4B). Consistently, heightened expression of SIINFEKL-bound H-2Kb and higher capacity to prime OT-I T cell proliferation were found in tumor-MDSC from TUDCA-treated mice bearing Pan02-OVA-ZsGreen tumors (Figure S4C–D). Thus, our data suggest that PERK deletion or UPR mitigation transform MDSC into cells that prime CD8+ T cell immunity in tumor beds.
Figure 4. PERK deletion functionally reprograms tumor-MDSC.
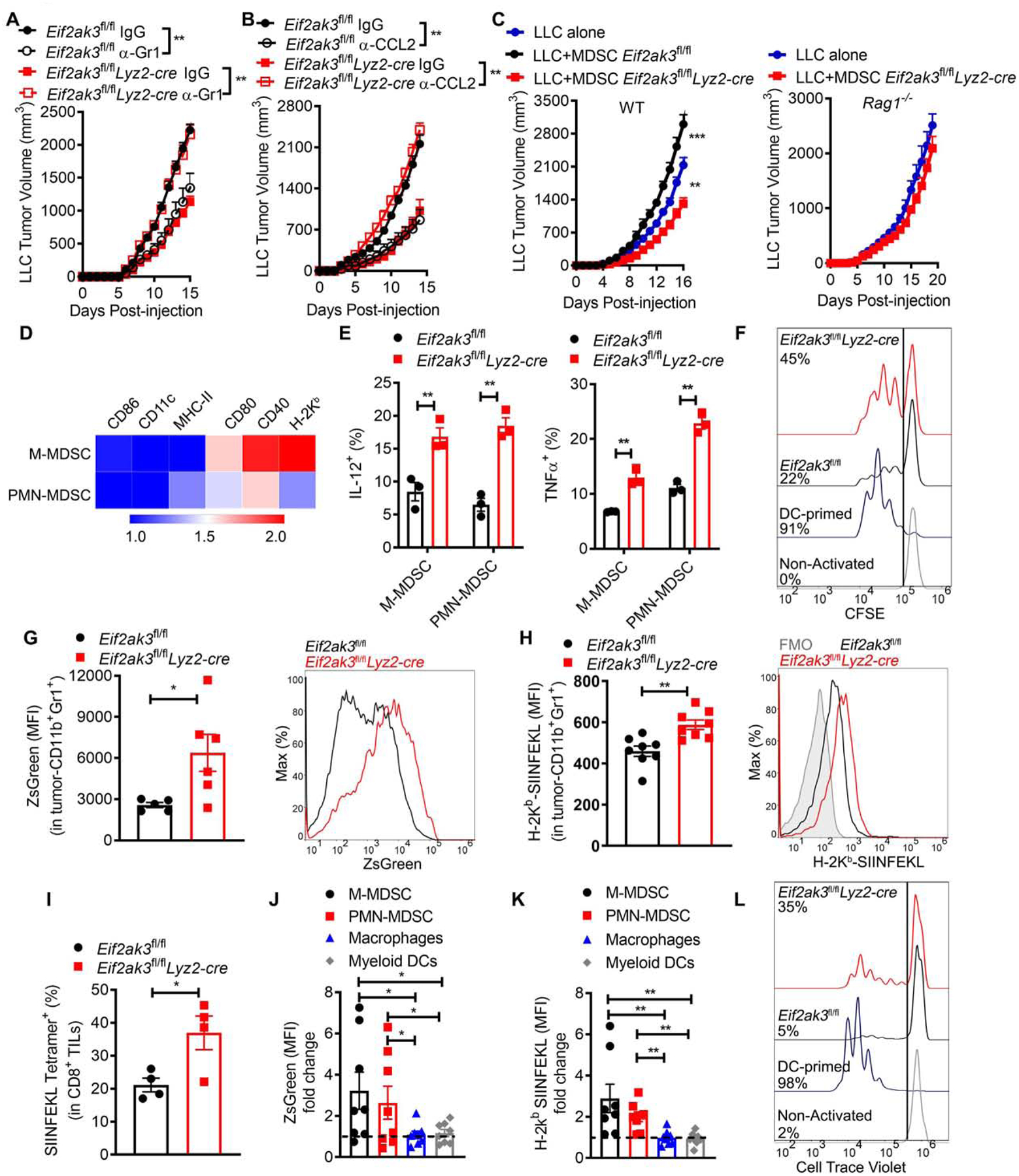
(A-B) LLC volume ± SEM in Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice treated every 3rd day with 250 μg anti-Gr1 (A), 250 μg anti-CCL2 (B) or IgG starting on the day of tumor injection. n=5.
(C) Tumor growth in WT (left) and Rag1−/− (right) mice injected with LLC cells alone or co-injected at a 1:1 ratio with LLC-tumor-MDSC from Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice. n=5.
(D) Relative expression heatmap of specific markers on tumor MDSC subsets from LLC-bearing Eif2ak3fl/flLyz2-cre mice compared to Eif2ak3fl/fl mice. n=10. Arbitrary units.
(E) Percentage of IL-12 (left) and TNF-α (right) in MDSC subsets from (D). n=3.
(F) Illustrative from 3 independent experiments showing proliferation of CFSE-labeled OT-I CD8+ T cells co-cultured with LLC-MDSC from Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice, pre-loaded with 1 mg/ml OVA (16 hours). Control OT-I cell proliferation was induced by OVA-loaded DCs.
(G-H) Merged MFI ± SEM (left) and sample histogram expression (right) of ZsGreen (G) and OVA-bound H-2Kb (H) in tumor-MDSC from Pan02-Ova-ZsGreen-bearing Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice. n=5 (G) and n= 8 (H).
(I) Percentage of SIINFEKL-H-2Kb-tetramer+ in CD8+ TILs from Pan02-Ova-ZsGreen tumors from Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice. n=4.
(J-K) Fold MFI change of ZsGreen (J) and SIINFEKL-bound H-2kb (K) in tumor-associated myeloid subsets from Eif2ak3fl/flLyz2-cre mice relative to controls. MFI values of specific myeloid subsets from PERK-null mice were divided over those from control mice. n=5
(L) Proliferation of cell-trace violet-labeled OT-I T cells co-cultured with tumor-MDSC (ratio 1:1/4) from (G). As positive control, OT-1 cells were co-cultured with GM-CSF plus IL-4-induced DCs loaded with OVA257–264 (1:1/4). Representative from 3 distinct repeats.
Statistics applied using one-way ANOVA or Student’s t-test, *, p<0.05; **, p<0.01. Please also see Figure S4.
PERK deletion transforms MDSC through NRF2 inhibition
To find the mediators by which PERK drives MDSC activity, we focused on CHOP, a downstream target of unmitigated cellular stress regulated by PERK, and previously linked to MDSC function (Thevenot et al., 2014). Reduced CHOP was detected in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice compared to MDSC from controls (Figure 5A). However, the decreased immunosuppressive activity found in PERK-null BM-MDSC treated with Thaps was not noted in Ddit3−/− BM-MDSC (Figure 5B). Also, ectopic expression of Ddit3 (Figure S5A) failed to restore immunosuppressive function of Eif2ak3−/− BM-MDSC after treatment with Thaps, while it enhanced the regulatory potential of vehicle-treated MDSC (Figure 5C). These results ruled out the impact of a reduced CHOP expression in the ablated immunosuppressive activity of PERK-deficient MDSC.
Figure 5. Functional switch of PERK-null MDSC occurs through impaired NRF2 signaling.
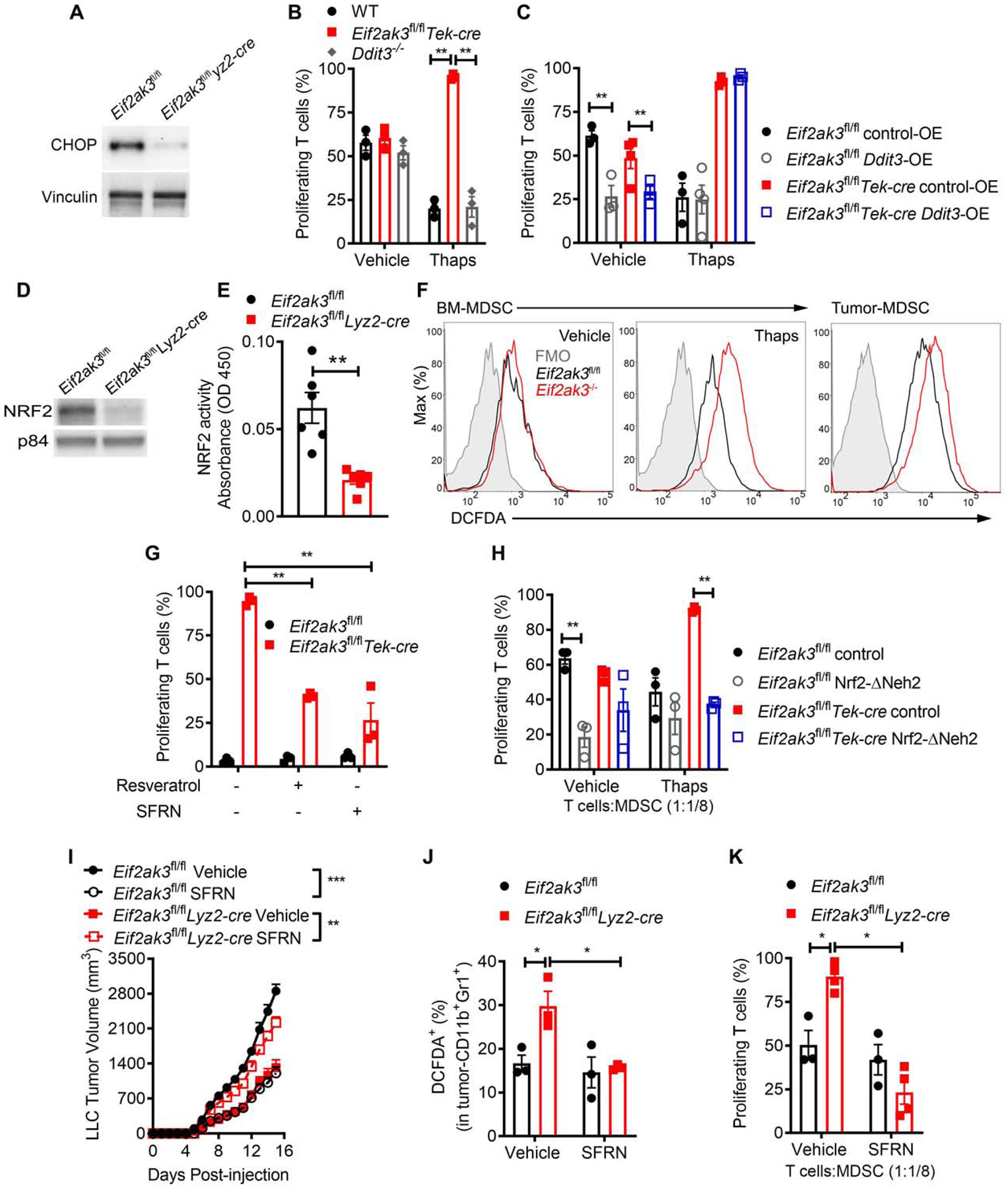
(A) Immunoblot of CHOP in tumor-MDSC from Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice bearing LLC tumors. Sample of n=5 distinct tumors.
(B) Proliferation of primed CFSE-labelled T cells co-cultured with wild type (WT), Eif2ak3−/− or Ddit3−/− BM-MDSC pre-treated or not with Thaps (24 hours) (ratio 1:1/8). n=3.
(C) Control and Eif2ak3−/− BM-MDSC were transduced with lentivirus expressing Ddit3 (Ddit3-OE) or control (control-OE), after which they were treated with or without Thaps (24 hours) and co-cultured with primed-T cells (ratio 1/8:1). T cell proliferation assessed as in (B). n=3.
(D-E) NRF2 immunoblot (D) and NRF2 binding to a consensus DNA-binding sequence (E) using 15 μg nuclear extracts from tumor-MDSC from LLC-bearing Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice. n=3 from distinct experiments.
(F) DCFDA in control MDSC (left), Thaps-treated BM-MDSC (middle), or tumor-MDSC (right) from controls and PERK-null mice. Illustrative result from 3 distinct experiments.
(G) Proliferation of activated T cells co-cultured for 72 hours with control or Eif2ak3−/− BM-MDSC pre-treated for 3 hours with vehicle, Resveratrol, or Sulforaphane (SFRN) and Thaps for 24 hours. n=3
(H) Proliferation of primed T cells co-cultured with BM-MDSC transduced with lentivirus coding for NRF2-ΔNeh2 or control and treated with Thaps (24 hours). n=3.
(I) Tumor Volume ± SEM in Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice bearing LLC-tumors and treated with vehicle or SFRN n=5.
(J) Percentages of DCFDA+ MDSC in tumors from (I). n=3.
(K) Percentage of proliferating T cells co-cultured with MDSC (ratio 1:1/4) from tumors from (I). n=4.
Statistics were applied using one-way ANOVA or student’s t-test, *, p<0.05; **, p<0.01, ***, p<0.001. Please also see Figure S5.
In light of the described phosphorylation of NRF2 by PERK in tumor cells (Cullinan et al., 2003), we next studied the mechanistic interaction between PERK and NRF2 in MDSC. Similar to the PERK upregulation in tumor-MDSC (Figure 1B), we noted higher nuclear NRF2 expression in tumor-MDSC from LLC-bearing mice, compared to splenic MDSC or iMC (Figure S5B). In addition, deletion of Nfe2l2 reduced the suppressive activity of tumor-MDSC to a similar extent as that induced by PERK elimination (Figure S5C). Also, consistent with the upstream role of PERK on NRF2 signaling, reduced expression of nuclear NRF2, lower binding of NRF2 to a consensus DNA sequence, and elevated ROS were found in tumor-MDSC and Thaps-treated BM-MDSC lacking PERK, compared to controls (Figure 5D–F, Figure S5D–E). Next, we validated the relevance of a thwarted NRF2 signaling in the lower regulatory function of Thaps-treated Eif2ak3−/− MDSC. Treatment with the indirect NRF2-inducing agents, Resveratrol or Sulforaphane (SFRN), partially restored the potential of Thaps-treated Eif2ak3−/− BM-MDSC to impair T cell proliferation (Figure 5G). Consistently, enforced expression of NRF2-ΔNeh2, a dominant active form of NRF2 (Shin et al., 2007) (Figure S5F), restored the immunosuppressive activity and lowered ROS content in Eif2ak3−/− BM-MDSC treated with Thaps, compared to the same cells carrying a control vector (Figure 5H, Figure S5G). Also, treatment with SFRN overcame the anti-tumor effects found in Eif2ak3fl/flLyz2-cre mice, while blunting tumor growth in Eif2ak3fl/fl mice (Figure 5I). Moreover, treatment of LLC-bearing Eif2ak3fl/flLyz2-cre mice with SFRN reduced ROS amounts and restored the immunoregulatory potential of tumor-MDSC, without affecting controls (Figure 5J–K). These results support the role of a reduced NRF2 activity in the effects found in PERK-deficient MDSC.
PERK regulates mitochondrial homeostasis in MDSC through NRF2
We elucidated whether the deletion of PERK impacted mitochondrial function in tumor-MDSC. Altered mitochondria morphology, diminished mitochondrial mass, and disrupted mitochondrial membrane potential were found in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice, compared to controls (Figure 6A–C). Also, in comparison to wild type MDSC, tumor-MDSC or Thaps-treated BM-MDSC from PERK-null mice had a lower mitochondrial respiratory activity, as suggested by a reduced oxygen consumption rate (OCR) upon mitochondrial stress studies (Figure 6D, Figure S6A). However, no changes in the extracellular acidification rate (ECAR) were found between tumor-MDSC from Eif2ak3fl/flLyz2-cre and control mice after glycolysis stress profiles (Figure S6B), indicating that PERK deletion altered mitochondrial function rather than glycolysis. Next, we identified the role of a decreased NRF2 in the mitochondrial alterations found in PERK-null MDSC. Enforced NRF2-ΔNeh2 expression restored mitochondrial respiratory activity, as tested by OCR, in Eif2ak3−/− BM-MDSC treated with Thaps (Figure 6E), without inducing variations in vehicle-treated MDSC (Figure S6C). Moreover, tumor-MDSC from LLC-bearing Eif2ak3fl/flLyz2-cre mice treated with SFRN had elevated OCR, compared to tumor-MDSC from untreated counterparts or from treated or untreated Eif2ak3fl/fl mice (Figure 6F). Thus, lower NRF2 signaling promotes mitochondrial respiratory dysfunction in PERK-ablated MDSC.
Figure 6. NRF2 regulates mitochondrial homeostasis in PERK-null MDSC.
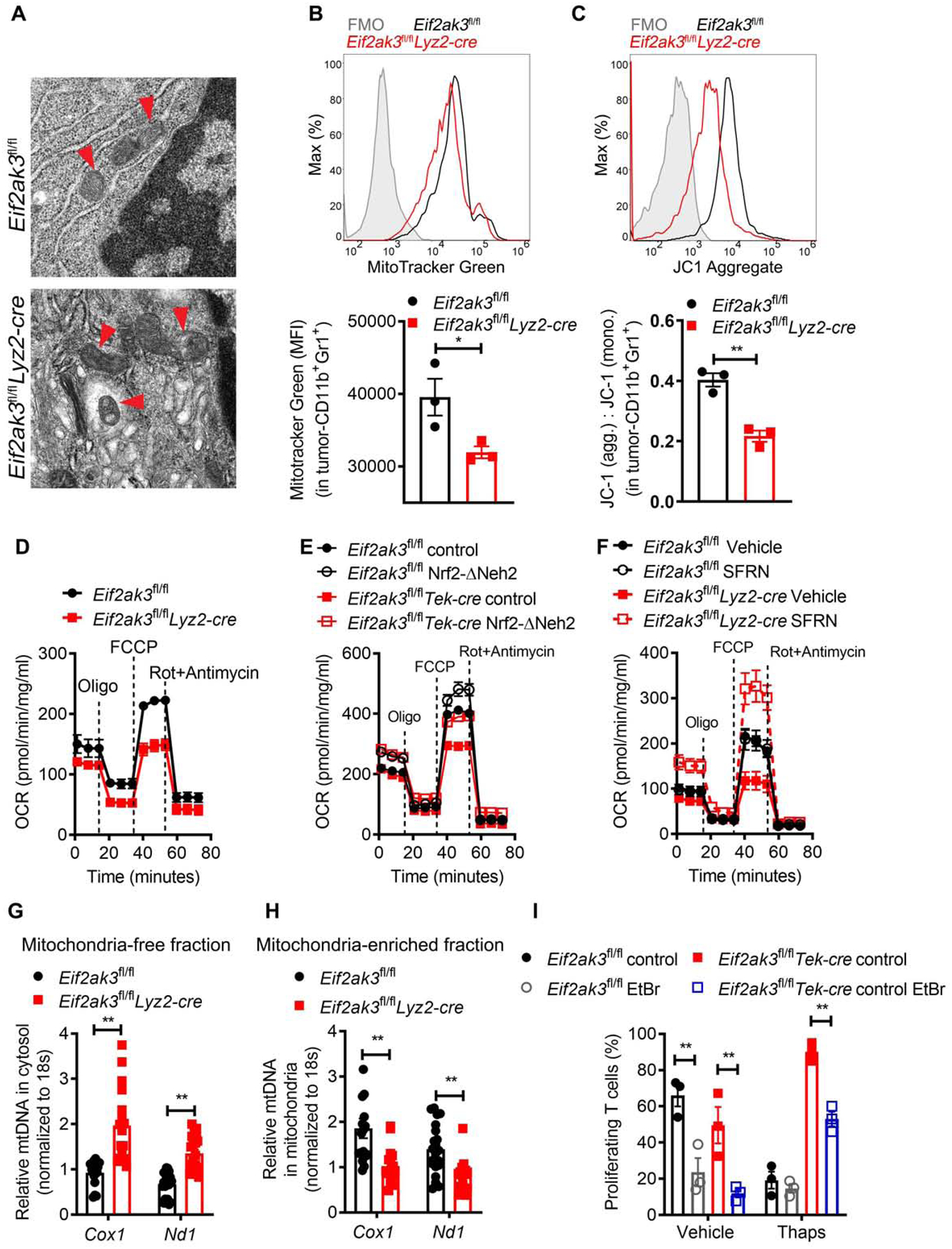
(A) Transmission electron microscopy image of tumor-MDSC from LLC-bearing Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice. Arrow heads point to mitochondria. Scale bar, 500 nm. Representative result from 3 independent experiments.
(B) Illustrative histogram of 3 distinct repeats (upper) and merged MFI values (lower) of Mitotracker fluorescence in tumor-MDSC from (A).
(C) Histogram of aggregated JC-1 fluorescence (upper) and JC-1 aggregates: JC-1 monomers ratio (upper) in tumor-MDSC from (A). n=3 independent repeats.
(D) Oxygen consumption rate (OCR) after mitochondrial stress analysis in tumor-MDSC from LLC-bearing Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice. n=5.
(E) OCR as in (D) in control and Eif2ak3−/− BM-MDSC transduced with NRF2-ΔNeh2 or control carrying lentivirus and treated with Thaps (24 hours). n=3.
(F) OCR as in (D) in tumor-MDSC from Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice bearing LLC tumors and treated with vehicle or SFRN. n=3.
(G-H) Relative Cox1 and Nd1 DNA expression in mitochondria-free cytosolic fractions (G) and mitochondrial-enriched fractions (H) from tumor-MDSC from (A). Results are triplicates of 5 samples/group.
(I) Proliferation of T cells co-cultured with Control and Eif2ak3−/− BM-MDSC developed in the presence of 150 ng/ml of EtBr and treated with or without Thaps (24 hours). n=3.
Statistics were applied using student’s t-test, *, p<0.05; **, p<0.01. Please also see Figure S6.
Because of the emerging role of the cytosolic mtDNA as a mediator of innate immunity (West et al., 2015) and the contribution of NRF2 signaling in maintaining mitochondrial integrity, we then assessed the presence of the mtDNA genes, Cox1 and Nd1, in mitochondria enriched and depleted cellular extracts from tumor-MDSC. Elevated mtDNA in mitochondrial-free cytosolic extracts and reduced mtDNA in mitochondrial enriched extracts were detected in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice compared to controls (Figure 6G–H). Similarly, PERK deficiency induced mtDNA accumulation in mitochondrial-free cytosolic extracts from BM-MDSC treated with Thaps, but not from non-treated BM-MDSC (Figure S6D–E). Moreover, depletion of mtDNA with ethidium bromide (EtBr) (Wang et al., 2018) partially restored suppressive activity of Eif2ak3−/− BM-MDSC treated with Thaps (Figure 6I), supporting the role of the amplified cytosolic mtDNA in the reprogramming of UPR-active PERK-deficient MDSC.
STING-dependent induction of Type I IFN drives effects of PERK ablation
The presence of mtDNA in the cytosol promotes STING pathway stimulation (Sliter et al., 2018). Consistently, heightened STING signaling, as indicated by a higher phosphorylation of TBK1 and nuclear IRF3, was detected in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice compared to controls (Figure 7A). Next, we identified the role of STING in the anti-tumor effects induced by PERK deletion, using myeloid cell PERK and STING null mice (Eif2ak3fl/flTmem173fl/flLyz2-cre). Dual elimination of PERK and STING in myeloid cells restored tumor growth and ability of tumor-MDSC to blunt T cell proliferation, compared to Eif2ak3fl/flLyz2-cre or Tmem173fl/flLyz2-cre mice (Figure 7B–C). Also, PERK-STING-deficient mice showed lower infiltration of total and IFNγ-producing CD8+ TILs compared to Eif2ak3fl/flLyz2-cre mice (Figure 7D, Figure S7A). Moreover, deletion of STING in PERK-null mice bearing Pan02-OVA-ZsGreen tumors limited the expansion of anti-SIINFEKL T cell responses and ablated the capacity of tumor-MDSC to induce ex vivo proliferation of OT-I CD8+ T cells (Figure 7E–F). Furthermore, consistent with STING activation, we found elevated expression of Type I IFN-related transcripts, Ifnb1, Isg15, Ifit3, and Cxcl10, in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice, which was attenuated after STING deletion (Figure 7G). The increased Ifnb1 mRNA noticed in tumor-MDSC from Eif2ak3fl/flLyz2-cre mice was not found in macrophages or myeloid DCs, indicating a preferential effect on MDSC (Figure S7B). Next, we tested the role of the Type I IFN in the anti-tumor effects noted in Eif2ak3fl/flLyz2-cre mice. Antibody-based blockade of IFNα/β receptor subunit 1 (IFNAR1) restored tumor growth in Eif2ak3fl/flLyz2-cre mice compared to IgG-treated counterparts (Figure 7H). Thus, activation of STING-driven Type I IFN drives the anti-tumor effects induced by myeloid cell-PERK deletion.
Figure 7. STING-dependent Type I IFN regulates functional switch of PERK-null MDSC.
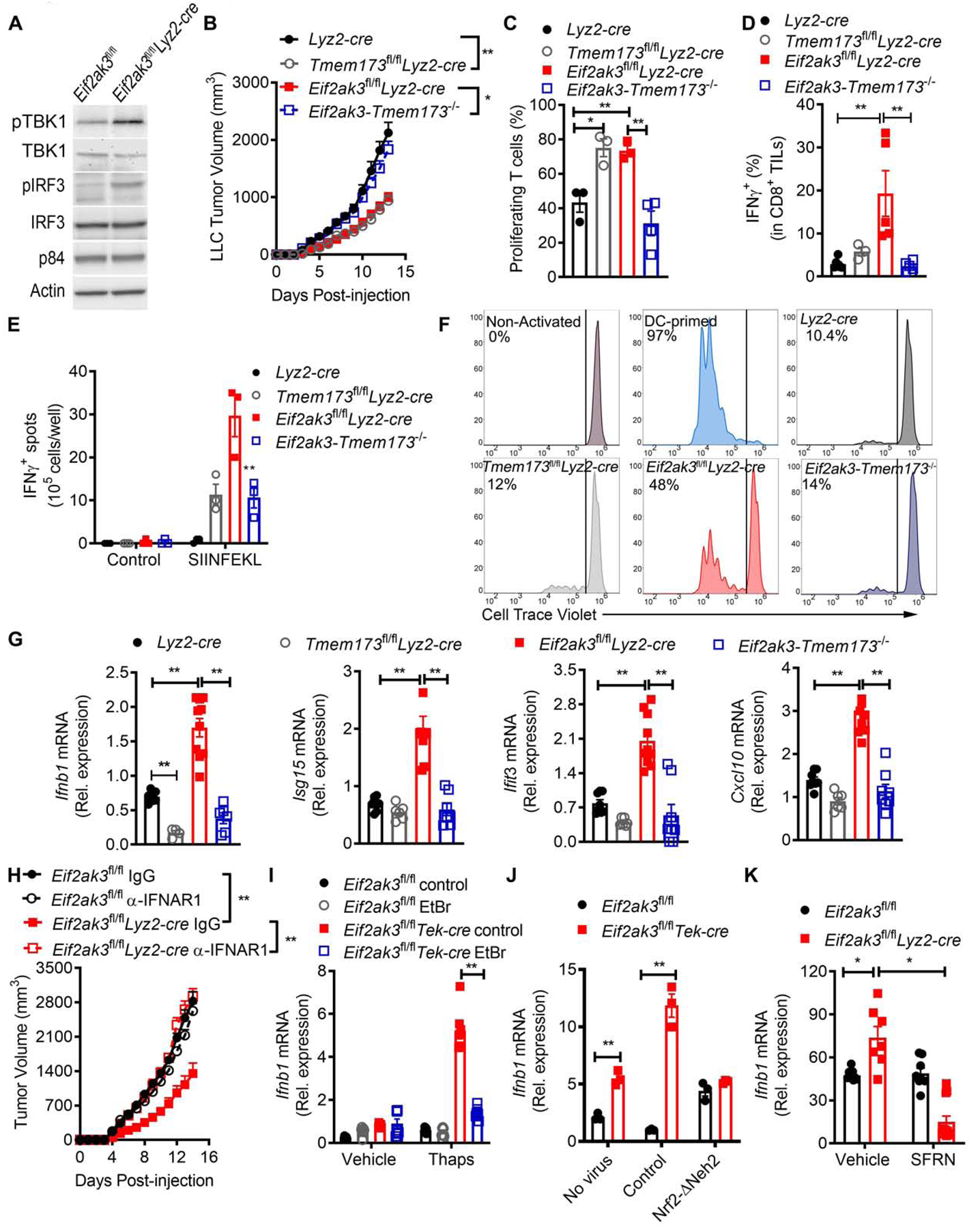
(A) Illustrative immunoblot from 3 independent experiments showing cytosolic phospho-TBK1 (pTBK1) and total TBK1, or nuclear phospho-IRF3 (pIRF3) and IRF3 in LLC-tumor MDSC from Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice.
(B) Tumor Volume ± SEM in Lyz2-cre, Eif2ak3fl/flLyz2-cre, Tmem173 fl/flLyz2-cre, and Eif2ak3fl/fl Tmem173 fl/flLyz2-cre (Eif2ak3-Tmem173 −/−) mice bearing LLC tumors for 15 days. n=7.
(C) Proliferation of primed CFSE-labeled T cells cultured with tumor-MDSC from (B) (ratio 1:1/4). n=3.
(D) Percentage of IFNγ+ in CD8+ TILs from (B). n=4.
(E) Tumor-draining lymph nodes from Lyz2-cre, Eif2ak3fl/flLyz2-cre, Tmem173 fl/flLyz2-cre and Eif2ak3-Tmem173 −/− mice bearing Pan02-Ova-ZsGreen tumors for 14 days were tested for IFNγ by EliSpot upon OVA257–264 activation. n=3/group.
(F) Proliferation of cell-trace violet-labeled OT-I CD8+ T cells co-cultured with tumor-MDSC from mice from (B). Sample of 5 independent repeats.
(G) Relative expression of Infb1, Ifit3, Cxcl10, mRNA in tumor-MDSC from (B). Duplicates from 5 samples/group.
(H) Tumor volume ± SEM in LLC-bearing Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice treated or not every 3 days with 1 mg anti-IFNAR1 antibody from day of tumor injection. n=5.
(I) Relative Ifnb1 mRNA expression in control and Eif2ak3−/− BM-MDSC developed in the presence of 150 ng/ml of EtBr and treated with Thaps (24 hours). n= 3.
(J) Relative Ifnb1 mRNA in control and Eif2ak3−/− BM-MDSC transduced with lentivirus coding for NRF2-ΔNeh2 or control and then treated with Thaps (24 hours).
(K) Relative Ifnb1 mRNA expression in LLC tumor-MDSC from Eif2ak3fl/fl and Eif2ak3fl/flLyz2-cre mice treated with vehicle or SFRN.
Statistics were applied using one-way ANOVA, *, p<0.05; **, p<0.01. Please also see Figure S7.
To define the mediators by which PERK deletion triggered the production of Type I IFN, we tested the impact of the amplified cytosolic mtDNA induced by reduced NRF2 signaling. Depletion of cytosolic mtDNA with EtBr prevented the Ifnb1 mRNA induction in Eif2ak3−/− BM-MDSC treated with Thaps (Figure 7I). Moreover, similar to the effects found in Eif2ak3−/− MDSC, higher cytosolic mtDNA and Ifnb1 mRNA were detected in tumor-MDSC from Nfe2l2−/− mice compared to those from wild type mice (Figure S7C–D). Consistently, NRF2-ΔNeh2 overexpression prevented the induction of Ifnb1 mRNA in Thaps-treated Eif2ak3−/− BM-MDSC, compared to controls (Figure 7J). Also, tumor-MDSC from Eif2ak3fl/flLyz2-cre mice treated with SFRN showed reduced Ifnb1 mRNA, compared to those from untreated counterparts (Figure 7K). Overall, the results show the crosstalk between reduced NRF2 signaling, cytosolic mtDNA, and STING-dependent production of Type I IFN as mediators for the effects induced by PERK deletion in MDSC.
Discussion
Here, we elucidated the role of PERK in the immunosuppressive polarization of MDSC in tumors. PERK deletion in MDSC reduced NRF2 signaling, which promoted STING-driven production of Type I IFN through amplified cytosolic mtDNA. Our data suggest a potential strategy to reprogram immunosuppressive myelopoiesis in tumors, which enhances the effects of cancer immunotherapy.
Although transient and moderate activation of UPR-associated signals support the physiological differentiation and function of immune cells (Bettigole et al., 2015; Dong et al., 2019; Iwakoshi et al., 2007; Martinon et al., 2010; Reimold et al., 2001), it has become evident that sustained and maladaptive priming of the UPR drivers promotes immune dysfunction (Condamine et al., 2014; Cubillos-Ruiz et al., 2015; Thevenot et al., 2014; Yan et al., 2016). In agreement with the immunoregulatory effect of the UPR, our results revealed a crucial role of PERK in the activity of MDSC, which was only manifested after exposure to the TME or stimulation of the UPR. In cancer cells undergoing unmitigated ER stress, the activation of PERK and IRE1α induces opposite and coordinated signals that control apoptotic vs. survival cell fate, respectively (Chang et al., 2018; Lu et al., 2014). Conversely, PERK deletion in tumor-MDSC did not correlate with alterations in the expression of phospho-IRE1α or apoptosis rates. The physiological activities of PERK have been mostly attributed to CHOP induction (Harding et al., 2000). However, the actions induced by PERK-null MDSC occurred in a CHOP-independent manner. Instead, PERK deletion transformed tumor-MDSC through NRF2 signaling inhibition, which supports previous reports in cancer cells showing the crosstalk between PERK and NRF2 in the survival to elevated ROS (Cullinan and Diehl, 2004). Rather than regulation of survival, our data showed that NRF2 signaling inhibition in PERK-deficient MDSC triggered ROS accumulation and thwarted mitochondrial respiratory function. In agreement, a recent study has suggested the relevance of the elevation of ROS in the reprogramming of M-MDSC into CD103+ DCs (Sharma et al., 2018). Conversely, previous reports have indicated the major role of the production of ROS in the immunoregulatory activity of MDSC (Corzo et al., 2009; Raber et al., 2014), indicating the paradoxical effects of the regulation of ROS in the functionality of tumor-MDSC.
STING-mediated Type I IFN production in DCs incites anti-tumor immunity (Corrales et al., 2015; Deng et al., 2014; Woo et al., 2014). Consistently, elimination of STING overcame the immune stimulatory effects triggered by PERK deletion in tumor-MDSC. Induction of STING-driven Type I IFN in PERK-deficient tumor-MDSC was the result of a lowered NRF2 signaling, which triggered accumulation of cytosolic mtDNA, an emerging activator of STING pathway (West et al., 2015). Deletion of PERK promoted the expression of Type I IFN in tumor-MDSC, but not in macrophages or myeloid DCs, which could be related with the active degradation of cytosolic mtDNA in macrophages (Xu et al., 2017) and the inefficient PERK deletion in myeloid DCs from Lyz2-cre mice. Although our data showed the promotion of STING-driven Type I IFN by reduced NRF2 through cytosolic mtDNA, an alternative negative effect of NRF2 on Tmem173 mRNA stability has been reported (Olagnier et al., 2018). Furthermore, although STING regulated the transformation of PERK-null MDSC, we also observed that myeloid cell-STING promoted tumor growth in PERK-competent mice. The pro-tumor vs. anti-tumor roles of STING can be argued in the light of reports that illustrated the paradoxical effects of STING in tumors. STING promotes effective anti-tumor immunity against highly immunogenic tumors (Woo et al., 2014), but also drives tumor growth in non-immunogenic tumor models (Lemos et al., 2016). The magnitude of the STING activation could be another explanation. STING promotes inflammation-driven carcinogenesis and immune tolerance towards spontaneously dying cells (Ahn et al., 2017; Ahn et al., 2014), whereas it activates anti-tumor immunity following amplified cell death induced by irradiation or ROS (Carroll et al., 2016; Deng et al., 2014). An additional possibility for the pro-tumor effects of STING in PERK-competent mice is the occurrence of STING-to-Type I IFN uncoupled events. In fact, PERK can directly limit Type I IFN responses by induction of phosphorylation-driven degradation of IFNAR1 (Bhattacharya et al., 2013). Also, STING regulates the activity of NFkB, a key driver of tumor-MDSC function (Sierra et al., 2017). Thus, studies elucidating the role of STING-to-Type I IFN dependent vs. independent actions in MDSC could clarify paradoxical effects and enable strategies to reinvigorate anti-tumor immunity.
The immunoregulatory role of the sustained UPR is not restricted to myeloid cells. TME-related stimulation of IRE1a, PERK, or CHOP promotes dysfunction of intra-tumoral T cells (Cao et al., 2019; Hurst et al., 2019; Song et al., 2018). Furthermore, PERK deletion in cancer cells impairs their adaptation to hypoxia, DNA damage, nutrient starvation, and high ROS, resulting in delayed tumor growth (Maas and Diehl, 2015; Wu et al., 2015). As such, inhibition of PERK has emerged as a promising cancer therapy (Atkins et al., 2013). However, the interest towards the use of PERK inhibitors has been affected by the reported pancreatic toxicity, which occurs in a Type I IFN-dependent manner (Yu et al., 2015). Our results showed that short-term treatment with low dose PERK inhibitors promoted anti-tumor protective immunity and boosted the effects of anti-PD-L1, without inducing toxicity. Despite these encouraging data, further research evaluating the activity of PERK inhibitors is warranted, as we anticipate that the dose, treatment period, route, and bioavailability could regulate their therapeutic vs. toxic effects. Alternatively, therapeutic use of TUDCA could mitigate UPR and induce dual anti-tumor and immunostimulatory effects in tumor-bearing hosts, without known toxicity. This remains to be tested in clinical studies and could have a major impact in the improvement of cancer immunotherapy.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
The authors declare that all the results supporting the findings of this study are available within the paper and its Supplemental Figures. Further information and requests for supporting data and resources should be directed to and will be fulfilled upon reasonable request by the Lead Contact, Paulo C. Rodriguez, PhD ([email protected]).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Experiments using mice were developed through an approved Institutional Animal Care and Use Committee (IACUC) protocol (IS00004043) and an active Institutional Biosafety Committee (IBC) study (#1385), both reviewed by the Integrity and Compliance board at the University of South Florida and Moffitt Cancer Center. Thus, the presented work has complied with all the relevant ethical regulations for animal testing and research. Wild type C57BL/6J mice (6 to 8 weeks) were from Envigo (Huntingdon, UK). Rag1−/− mice (NOD.129S7 (B6)-Rag1tm1Mom/J), Lyz2-cre mice (B6.129P2-Lyz2tm1(cre)Ifo/J), Tek-cre mice (B6.Cg-Tg(Tek-cre)1Ywa/J), Eif2ak3fl/fl mice (Eif2ak3tm1.2Drc/J), Nfe2l2−/− mice (B6.129X1-Nfe2l2tm1Ywk/J), tdTomato reporter mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J), and OT-I mice (C57BL/6-Tg (Tcra-Tcrb) 1100Mjb/J) were from the Jackson laboratories (Bar Harbor, ME). Tmem173fl/fl mice were a kind gift from Dr. John C. Cambier (University of Colorado Denver and National Jewish Health). LSL-KrasG12D/+Trp53fl/fl mice were developed in (Rutkowski et al., 2015). Eif2ak3fl/flLyz2-cre mice were created after breeding Eif2ak3fl/fl mice with Lyz2-cre mice; while BM-MDSC were obtained from Eif2ak3fl/fl and Eif2ak3fl/flTek-cre mice. Eif2ak3fl/flTmem173fl/flLyz2-cre mice were developed after breeding Eif2ak3fl/fl, Tmem173fl/fl, and Lyz2-cre mice. Tmem173fl/flLyz2-cre mice were developed after breeding Tmem173fl/fl and Lyz2-cre mice. tdTomatofl/+Lyz2-cre and tdTomatofl/+Tek-cre reporter mice were developed after crossing Lyz2-cre or Tek-cre mice with tdTomatofl/fl mice. Mice of the same sex were randomly assigned to all experimental cohorts. All mice were maintained under specific pathogen-free conditions and used at 6–10 weeks of age.
Human Materials
All human studies were covered through the approved Institutional Review Board (IRB) exempt protocol #19223, previously reviewed by the Regulatory Affairs Committee Board at Moffitt Cancer Center. Human peripheral blood mononuclear samples (PBMC) were obtained from One-Blood, Tampa, FL. Human bone marrow stem cells were from a repository established by Dr. Pilon-Thomas (Moffitt Cancer Center). Both males and females were included in the donor pools and sample information maintained anonymous. All de-identified patients signed approved consent forms. Also, Met-NSCLC and advanced high-grade serous ovarian cancer tissue microarrays (TMA) were available from the Moffitt Cancer Center Biorepository.
Cell Lines
Lewis lung carcinoma (LLC; #CRL-1642), B16-F10 (#CRL-6475), and EG7 (#CRL-2113) were used for s.c. tumor models and obtained from the American Type Culture Collection (ATCC). Ovarian ID8-Defb29-Vegfa and Pan02-Ova-ZsGreen cells lines were provided by Dr. Jose Conejo-Garcia and Dr. Shari Pilon Thomas, respectively (Perales-Puchalt et al., 2017; Svoronos et al., 2017). HEK293T cells (#CRL-11268) were obtained from ATCC. All cell lines were validated to be mycoplasma-free using the Universal Mycoplasma Detection Kit (#30–1012K, ATCC), and cultured in RPMI-1640 supplemented with 2 mM L-glutamine, 10 mM HEPES, 150 U/ml streptomycin, 200 U/ml Penicillin, 20 μM β-mercaptoethanol and 10% heat-inactivated Fetal bovine serum (FBS), and maintained at 37°C in a humidified incubator with 5% CO2.
METHOD DETAILS
Tumor models
Mice were subcutaneously (s.c.) injected with LLC, B16-F10, EG7 or Pan02-Ova-ZsGreen and tumor volume assessed using calipers and calculated using the formula [(small diameter)2 × (large diameter) × 0.5]. For the ovarian carcinoma model; ID8-Defb29-Vegfa cells were injected intraperitoneal (i.p.) and body weight was assessed daily and mice euthanized when they gained 30% of their body weight. To develop soft tissue autochthonous flank sarcomas, mice with latent mutations in LSL-KrasG12D/+Trp53fl/fl mice were irradiated for two consecutive days with 550 rads, followed by reconstitution with bone marrow from Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice. Autochthonous flank sarcomas were then initiated 7 weeks later by intramuscular delivery of 2.5 × 108 plaque-forming units of adenovirus coding for Cre recombinase (Gene Transfer Vector Core, University of Iowa) (Rutkowski et al., 2015). PERK inhibitors AMG-44 (12 or 24 mg/kg), GSK-2606414 (25 mg/kg) were administered i.p. daily and starting at day 6 post-tumor implantation and until study endpoint. Furthermore, mice received i.p. TUDCA (250 mg/kg) or Thapsigargin (Thaps, 100 μg/kg) after tumors were established (day 6 post-LLC injection). To deplete CD8+ T cells, tumor-bearing mice were injected i.p. with 400 μg anti-CD8 antibody (clone 53–6.7, BioXcell) at day 0 followed by every 3rd day treatments until experimental endpoint. Same approach was applied for elimination of Gr1+ cells using anti-Gr1 antibody (250 μg/mouse, clone RB6–8C5, BioXcell), prevention of myeloid cells mobilization using anti-CCL2 (250 mg/mouse, clone 2H5, BioXcell), blockade of PD-L1 (250 μg/mouse, clone 10F.9G2, BioXcell), and neutralization of interferon type 1 receptor using anti-IFNAR1 (1 mg/mouse, clone MAR1–5A3, BioXcell). For NRF2 signaling induction, tumor-bearing mice were treated i.p. with D, L-Sulphoraphane (25 mg/kg, 3 days per week) starting at day 6 post-tumor injection.
Development of mouse and human MDSC from bone marrow (BM) precursors
Mouse BM-MDSC were generated after culturing BM precursors with GM-CSF and G-CSF (20 ng/ml each) for 4 days. In vitro generated MDSC were treated with either LLC tumor explant supernatants (TES, since day 0) or Thaps (200 nM, for the last 24 hours of culture). Human BM-MDSC were developed after culturing BM or peripheral blood precursors from healthy donors with GM-CSF and IL-6 (10 ng/ml each) for a period of 6 days (Thevenot et al., 2014). To induce UPR activation, MDSC were treated with either supernatants from the renal cell carcinoma cell line 786–0 (hTES) for 6 days (Rodriguez et al., 2009) or Thaps during the last 24 hours before collection (200 nM). In some experiments, MDSC were pre-incubated for 3 hours with Resveratrol (100 μM, Sigma-Aldrich), D,L-Sulphoraphane (20 μM), AMG-44 (5 μM), or TUDCA (500 nM) before treating with Thaps.
Tumor digestion
Minced tumors were digested with DNase I and Liberase (Roche USA, Branchburg, NJ). Tumor digests were then treated with ACK buffer (Ammonium-Chloride-Potassium) to lyse the red blood cells.
Cell isolation
CD3+ T cells were enriched using mouse or human T cell negative selection kits (MagniSort, Invitrogen) from the spleen and lymph nodes of C57BL/6 mice; or from purchased human buffy-coat units (One-Blood, Tampa, FL). Purity ranged between 95% and 99% as tested by flow cytometry. For antigen-specific priming studies, CD8+ T cells were enriched using negative selection kits from the spleens of OT-I mice. For ACT experiments, sorted CD8+ T cells were previously activated with OVA257–264 (SIINFEKL) for 48 hours before transferring to EG7-bearing mice. Tumor-MDSC were isolated from cellular suspensions of digested tumors followed by positive selection (MojoSort Streptavidin Nanobeads, Biolegend) using anti-Gr1 (Clone RB6–8C5, eBioscience) or anti-CD11b-biotinylated antibodies (Clone M1/70, StemCell Technologies, Vancouver). Also, M-MDSC (CD11b+Gr1+Ly6GlowLy6Chigh), PMN-MDSC (CD11b+Gr1+Ly6G+Ly6Clow), macrophages (CD11b+Gr1negF4/80+CD11cneg), and myeloid DCs (CD11b+Gr1negF4/80negCD11c+) were sorted from tumors in a FACSAriaII (BD Biosciences).
In vitro and ex vivo suppression assays
Murine BM-MDSC (ratio 1:1/8) or tumor-MDSC (ratio 1:1/4) were co-cultured with negatively selected CD3+ T cells in a 96 well plate bound with anti-CD3 and anti-CD28 (1 μg/ml each, eBioscience) for 3 days. T cell proliferation was assessed with Carboxyfluorescein succinimidyl ester (CFSE) or Cell-trace violet (Invitrogen) dye dilution detected by flow cytometry. For human studies, MDSC were co-cultured with enriched CD3+ T cells activated with soluble anti-CD3 (1 μg/ml, clone OKT3) and anti-CD28 (0.5 μg/ml, clone L293) for 3 days in a 96 well plate bound with goat anti-mouse IgG (10 μg/ml). Results are expressed as percentage of proliferating T cells, determined by the dilution of fluorescence compared with non-stimulated T cells.
Antigen presentation co-cultures
For antigen loading experiments, MDSC were sorted from LLC tumors and then pulsed with 1 μg/ml complete ovalbumin (OVA). After 16 hours, cells were washed twice and 2.5×104 MDSC co-cultured with 1×105 CFSE-labeled naïve CD8+ T cells from spleens of OT-I mice (ratio 0.25:1). Positive controls included co-cultures of OT-I T cells with GM-CSF plus IL-4-derived BM-DCs loaded with 1 μg/ml complete OVA. For cross-priming assays, 1.25×104 MDSC from Pan02-Ova-ZsGreen tumors were co-cultured with 5×104 naïve CD8+ T cells (1/4:1) from spleens of OT-I mice and labelled with Cell-trace violet. Positive control wells included OT-I T cells co-cultured with GM-CSF plus IL-4-derived BM-DCs loaded with 1 μg/ml OVA257–264 peptide. T cell proliferation was measured after 3 days of co-culture.
Cloning of pHIV-OVA (deltaN)-2A-ZsGreen
The lentiviral constitutive expression vector pHIV-OVAdeltaN-2A-ZsGreen was created by cloning chicken ovalbumin lacking the sequence coding for amino acids 1–49 from the vector pcDNA3-deltaOVA (Addgene 64595) into the pHIV-T2A-ZsGreen vector backbone, which was generated from the pHIV-IRES-ZsGreen (Addgene 18121) by replacing the IRES sequence with the self-cleaving 2A sequence. Cloning reactions were performed according to manufacturer’s recommendation using the InFusion-HD Cloning Plus kit from TakaraBio (#638909). Lentivirus containing supernatant was produced by transfecting pVSV-G, pSPAX2 and pHIV-OVA (deltaN)-2A-ZsGreen into LentiX-293T (TakaraBio). Collected supernatants were filtered (0.45 mm; ThermoFisher F2500-5) and lentivirus concentrated by ultracentrifugation at 24000 rpm for 2 hours at 4° C.
Overexpression vectors and transduction of MDSC
Lentivirus expressing Ddit3 was created by inserting the full-length Ddit3 cDNA into the lentiviral vector pCDH-MSCV-MCS-EF1α-GFP. Similarly, NRF2-ΔNeh2, a dominant active form of NRF2 highly resistant to proteasome degradation (Shin et al., 2007), was inserted in the same lentiviral vector. Empty vectors were used as negative controls. For virus production, 293T-cells (#CRL-3216, ATCC) were co-transfected with the lentiviral expression vector and a packaging vector that expresses the lentiviral envelope; and viral supernatants collected 48–96 hours post-infection. For lentiviral transduction, mouse BM precursors were incubated with GM-CSF and G-CSF (20 ng/ml each) and 24 hours later, MDSC transduced in the presence of 5 μg polybrene transfection reagent (Millipore) for 3 days until endpoint to perform readouts.
Adoptive and Co-Transfer experiments
For adoptive T cell therapy (ACT), mice bearing OVA+ EG7 tumors for 6 days received daily doses of TUDCA or vehicle until experimental endpoint. Additionally, specific cohorts of mice received on day 7 post-tumor injection, 1×106 CD8+ OT-I T cells sorted from OT-I splenocytes previously activated with 1 μg/ml OVA257–264 for 48 hours. In MDSC co-injection experiments, 1×106 tumor MDSC from Eif2ak3fl/fl or Eif2ak3fl/flLyz2-cre mice bearing LLC tumors were co-injected s.c. with 1×106 LLC cells in 1:1 ratio into C57BL/6J mice or Rag1−/− mice.
IFNγ ELISpot
Spleens from EG7-bearing mice undergoing or not ACT were collected 5 days after T cell transfer and 1×105 splenocytes seeded in ELISpot plates containing pre-bound IFNγ capturing antibody in the presence or the absence of 1 μg/ml OVA257–264. Also, tumor draining lymph nodes from Pan02-Ova-ZsGreen-bearing mice were collected at endpoint and 1×105 cells seeded in ELISpot plates containing pre-bound IFNγ capturing antibody in the presence or the absence of 1 mg/ml OVA257–264. Production of IFNγ was detected 24 hours later by ELISpot using Mouse IFNγ ELISpot (eBioscience).
Flow cytometry staining
The conjugated antibodies and probes used for flow cytometry are listed in the Key Resources Table. For surface staining, cells were labelled with the appropriate antibodies in the presence of Fc blocker. For intracellular staining, surface-labeled cells were fixed with Cytofix/Cytoperm™ Solution (BD Biosciences), washed in Perm/Wash™ 1X solution, and labelled with intracellular antibodies. Cells were then washed in Perm/Wash™ 1X and PBS. Live vs. dead cell discrimination was performed prior to antibody labeling by Zombie Fixable Viability dye (Biolegend). Ex vivo intracellular staining for IL-12 and TNFα was performed on isolated cells after stimulation for 6 hours with LPS (1 μg/ml, Sigma Aldrich) in the presence of Golgi stop (0.8 ml/ml, BD Biosciences). For IFNγ staining, cells were incubated for 5 hours with phorbol myristate acetate (PMA, 750 ng/mL, Sigma Aldrich) and ionomycin (50 μg/mL, Sigma-Aldrich) in the presence and Golgi stop (0.8 μl/ml). For ER-tracker and Mitotracker staining, cells were probed with 100 nM of ER tracker green or 200 nM of Mitotracker green (Invitrogen) and then stained for surface markers. For Mitochondrial membrane potential, cells were stained with JC-1 flow cytometry assay kit (Cayman chemicals) followed by surface markers staining. ROS were detected by DCFDA (10 μM) or Dihydroethidium (DHE, 10 μM). Data acquisition was performed in a CytoFLEX II (Beckman Coulter) or LSRII (BD Biosciences). All analysis was performed using FlowJo version 11 software.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 BV785 | Biolegend | Cat# 103149 AB_2564590 |
| Anti-mouse CD45 BV605 | Biolegend | Cat# 103140 AB_2562342 |
| Anti-mouse CD11b BV421 | Biolegend | Cat# 101236 AB_11203704 |
| Anti-mouse CD11b PECy7 | Biolegend | Cat# 101216 AB_312799 |
| Anti-mouse Gr1 PE/Dazzle 594 | Biolegend | Cat# 108452 AB_2564249 |
| Anti-mouse Ly-6C APC-Cy7 | Biolegend | Cat# 128025 AB_10643867 |
| Anti-mouse Ly-6C Pacific Blue | Biolegend | Cat# 128014 AB_1732079 |
| Anti-mouse Ly6G BV605 | Biolegend | Cat# 127639 AB_2565880 |
| Anti-mouse CD8 APC | Biolegend | Cat# 100712 AB_312751 |
| Anti-mouse CD44 FITC | Biolegend | Cat# 103022 AB_493685 |
| Anti-mouse F4/80 Alexa Flour 700 | Biolegend | Cat# 123130 AB_2293450 |
| Anti-mouse TNF-α PE | Biolegend | Cat# 506305 AB_315426 |
| Anti-mouse H-2Kb-SIINFEKL PE | Biolegend | Cat# 141604 AB_10895905 |
| Anti-mouse CD11c BV785 | Biolegend | Cat# 117336 AB_2565268 |
| Anti-mouse CD45 BUV395 | BD | Cat# 564279 AB_2651134 |
| Anti-mouse CD11b BV711 | BD | Cat# 563168 AB_2716860 |
| Anti-mouse CD11b PE | BD | Cat# 561001 AB_10563205 |
| Anti-mouse CD11b PerCpCy5.5 | BD | Cat# 550993 AB_394002 |
| Anti-mouse G-1 BUV395 | BD | Cat# 563849 AB_2738450 |
| Anti-mouse Gr1 PercpCy5.5 | BD | Cat# 552093 AB_394334 |
| Anti-mouse Gr1 APC | BD | Cat# 553129 AB_398532 |
| Anti-mouse Ly-6G BUV395 | BD | Cat# 563978 AB_2716852 |
| Anti-mouse CD8 PE | BD | Cat# 553033 AB_394571 |
| Anti-mouse CD8 PerCpCy5.5 | BD | Cat# 561109 AB_10563417 |
| Anti-mouse CD69 PE | BD | Cat# 553237 AB_394726 |
| Anti-mouse MHC I (H-2Kb) PE | BD | Cat# 553570 AB_394928 |
| Anti-mouse CD40 PE | BD | Cat# 553791 AB_395055 |
| Anti-mouse IL-12p40 PE | BD | Cat# 554479 AB_395420 |
| Anti-mouse CD3 APC | BD | Cat# 553066 AB_398529 |
| Anti-mouse Ly-6C FITC | BD | Cat# 553104 AB_394628 |
| Anti-mouse IFNγ APC | eBiosciences | Cat# 17-7311-82 AB_469504 |
| Anti-mouse CD45 Alexa Flour 700 | eBiosciences | Cat# 56-0451-82 AB_891454 |
| Anti-mouse Ly-6G APC | Tonbo | Cat# 20-1276-U100 AB_2621589 |
| Anti-mouse Tetramer/PE-H-2Kb SIINFEKL PE | MBL international | Cat# TB-5001–1 |
| H-2Db gp100 Tetramer-EGSRNQDWL-PE | MBL international | Cat# TS-M546–1 |
| PERK (C33E10) Rb mAb | Cell signaling | Cat# 3192 AB_2095847 |
| Phospho-PERK (Thr980) Rb mAb | Cell signaling | Cat# 3179 AB_2095853 |
| IRE1α (14C10) Rb mAb | Cell signaling | Cat# 3294 AB_823545 |
| Phospho TBK1/NAK (Ser172) (D52C2) XP Rb mAb | Cell signaling | Cat# 5483 AB_10693472 |
| TBK1/NAK (D1B4) Rb mAb | Cell signaling | Cat# 3504 AB_2255663 |
| Phospho IRF3 (Ser396) (D6O1M) Rb mAb | Cell signaling | Cat# 29047 AB_2773013 |
| IRF3 (D83B9) Rb mAb | Cell signaling | Cat# 4302 AB_1904036 |
| NRF2 (D1Z9C) XP Rb mAb | Cell signaling | Cat# 12721 AB_2715528 |
| Insulin (C27C9) Rb mAb | Cell signaling | Cat# 4590 AB_659820 |
| Alexa Flour 647 conjugated anti-rabbit | Cell signaling | Cat# 4414 AB_10693544 |
| Chop Rb polyAb | Santa Cruz | Cat# Sc-793 AB_631364 |
| Phospho IRE1α (S724) Rb polyAb | abcam | Cat# 48187 AB_873899 |
| Arginase Ms mAb | BD | Cat# 610709 AB_398032 |
| Nos2 Ms mAb | BD | Cat# 610432 AB_397808 |
| Anti-mouse CD3e | BD | Cat# 553058 AB_394591 |
| Anti-mouse CD28 | BD | Cat# 553294 AB_394763 |
| Anti-Human CD28 | BD | Cat# 348040 AB_400367 |
| Anti-Human CD3e | ThermoFisher | Cat# MA1–10176 AB_11157009 |
| InVivoMAb anti-mouse CD8α | BioXcell | Cat# BE0004–1 AB_1107671 |
| InVivoMAb anti-mouse IFNAR-1 | BioXcell | Cat# BE0241 AB_2687723 |
| InVivoMAb anti-mouse Ly6G/Ly6C (Gr1) | BioXcell | Cat# BE0075 AB_10312146 |
| InVivoMAb anti-mouse/human/rat CCL2 (MCP-1) | BioXcell | Cat# BE0185 AB_10950302 |
| InVivoMAb anti-mouse PD-L1 (B7–H1) | BioXcell | Cat# BE0101 AB_10949073 |
| Goat anti-mouse IgG | KPL | Cat# 01-18-06 |
| Oligonucleotides | ||
| qRT-PCR primers are referenced in Table S1 | See Table S1 | |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 Mice | Envigo | Cat# 044 |
| NOD.129S7 (B6)-Rag1tm1Mom/J | Jackson Laboratory | Cat# 003729 |
| Eif2ak3tm1.2Drc/J | Jackson Laboratory | Cat# 023066 |
| B6.129P2-Lyz2tm1(cre)Ifo/J | Jackson Laboratory | Cat# 004781 |
| B6.Cg-Tg(Tek-cre)1Ywa/J | Jackson Laboratory | Cat# 008863 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | Cat# 003831 |
| B6.129X1-Nfe2l2 tm1Ywk/J | Jackson Laboratory | Cat# 017009 |
| B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | Cat# 007909 |
| Tmem17fl/fl | Dr. John C. Cambier | N/A |
| LSL-KrasG12D/+Trp53fl/fl | PMID: 25533336 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse GM-CSF | Gemini | Cat# 300–308P |
| Mouse G-CSF | Gemini | Cat# 300–207P |
| Human GM-CSF | Invitrogen | Cat# BMS324 |
| Human IL-6 | Invitrogen | Cat# BMS341 |
| Tauroursodeoxycholic Acid | Calbiochem | Cat# 580549–5GM |
| AMG-PERK-44 | Tocris | Cat# 5517 |
| PERK Inhibitor I, GSK2606414 | Sigma Aldrich | Cat# 516535–5MG |
| D,L-Sulforaphane | Sigma Aldrich | Cat# 574215–25MG |
| Resveratrol | Sigma Aldrich | Cat# R5010–100MG |
| Thapsigargin | Sigma Aldrich | Cat# T9033–5MG |
| LPS | Sigma Aldrich | Cat# L5293–2ML |
| PMA | Sigma Aldrich | Cat# P1585–1MG |
| DAPI | Sigma Aldrich | Cat# D9542 |
| Ethidium Bromide | Sigma Aldrich | Cat# E8751 |
| Ionomycin | Sigma Aldrich | Cat# I3909–1ML |
| Golgi Stop | BD | Cat# 554724 |
| DNase I | Roche | Cat# 10104159001 |
| Liberase | Roche | Cat# 05401127001 |
| Ovalbumin residue (Ova257–264) | Anaspec peptide | Cat# 60193 |
| Critical Commercial Assays | ||
| TransAM NRF2 | Active Motif | Cat# 50296 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagent | ThermoFisher | Cat# 78833 |
| Mitochondrial Isolation Kit for Cultured Cells | ThermoFisher | Cat# 89874 |
| Verso cDNA synthesis Kit | ThermoFisher | Cat# AB-1453/B |
| Mito-Tracker Green | Invitrogen | Cat# M7514 |
| ER-Tracker Green | Invitrogen | Cat# E34251 |
| Cell Trace Violet | Invitrogen | Cat# C34557 |
| Vybrant CFDA SE Cell Tracer Kit | Invitrogen | Cat# V12883 |
| Zombie NIR Fixable Viability Kit | Invitrogen | Cat# 423105 |
| JC-1 Mitochondrial Membrane potential | Cayman | Cat# 701560 |
| iTaq universal SYBR green supermix | BIO-RAD | Cat# 1725121 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat# 69504 |
| Seahorse XFp Cell Mito Stress Test kit | Agilent Technologies | Cat# 103010–100 |
| Seahorse XFp Glycolysis Stress Kit | Agilent Technologies | Cat# 103017–100 |
| Experimental Models: Cell Lines | ||
| LLC | ATCC | CRL-1642 |
| B16-F10 | ATCC | CRL-6475 |
| E.G7-OVA | ATCC | CRL-2113 |
| ID8-Defb29-Vegfa | N.A | N.A |
| Pan02-Ova-ZsGreen | N.A | N.A |
| Deposited Data | ||
| N/A | ||
| Biological Samples | ||
| Human BM cells | Shari Pilon-Thomas, PhD | N.A. |
| TMA slides were from Tissue Core Facility | TMA slides were from Tissue Core Facility | TMA slides were from Tissue Core Facility |
| Blood buffy coat units | One-Blood, Tampa | |
| Bacterial and Virus Strains | ||
| Lentivirus | N.A | N.A |
| Adenovirus coding Cre Recombinase | Gene Transfer Vector Core (Univ. of Iowa) | N.A |
| Recombinant DNA | ||
| pCDH-MSCV-MCS-EF1α-GFP | N.A | N.A |
| Ddit3 | N.A | N.A |
| NRF2-ΔNeh2 | Dr. Thomas Kensler (Univ. of Pittsburgh) |
N.A |
| Software and Algorithms | ||
| Prism 7 | GraphPad | |
| FlowJO | FlowJo | |
| Wave | Agilent Technologies | |
| Aperio Image scope | Leica Biosystems | |
| Zen 2.3 (blue edition) | Carl Zeiss AG | |
| Definiens Tissue Studio software v4.7 | Definiens AG | |
| Halo | Indica labs | |
| Other | ||
Immunoblot Analysis
Equal protein amounts of total and nuclear cell lysates were electrophoresed in 8 or 10% Tris-Glycine gels (Novex-Invitrogen), transferred to PVDF membranes by iBlot™ Gel Transfer Device (ThermoFisher), and blotted with the corresponding primary and secondary antibodies described in Key Resources Table. For nuclear fraction isolation, cells were lysed using NE-PER Nuclear and Cytoplasmic extraction kit (ThermoFisher). Membrane-bound immune complexes were detected by ChemiDoc™ Imaging System (Bio-Rad, #17001401).
Quantitative PCR
Total RNA was isolated from MDSC using TRIzol (Life Technologies). Reverse transcription was performed using Verso cDNA Synthesis Kit (Thermo Scientific). Quantitative PCR reactions were prepared by using Bio-Rad SYBR green master mix and performed on an Applied Biosystems thermocycler (7900 HT) using primers described in Table S1.
NRF2 binding activity
Nucleic protein extracts (15 μg) from tumor-MDSC or in vitro-generated MDSC were monitored for binding of NRF2 to a DNA consensus sequence though a TransAM® Kit (Active Motif).
Seahorse metabolism assay
Oxygen consumption rate (OCR) through mitochondrial stress test and extracellular acidification rate (ECAR) by glycolysis stress profile were measured using a XF96 extracellular flux analyzer (Seahorse Bioscience). MDSC were plated onto CellTak pre-coated wells (1×105 cells/well in triplicate) and subjected to mitochondrial stress and glycolysis stress protocols using specific non-buffered XF base mediums. For OCR, cells were analyzed under basal conditions and in response to 2 μM oligomycin, 2 μM fluorocarbonyl-cyanide-phenylhydrazone (FCCP), and 0.5 μM rotenone. For ECAR, cells were plated in XF media lacking glucose and monitored under basal conditions and in response to 10 μM glucose, 10 μM oligomycin, and 100 mM 2-Deoxy-D-glucose (2-DG). Final values were obtained after protein normalization among the conditions.
Cellular fractionation and quantification of mtDNA
Cytosolic and mitochondrial cellular fractionation of BM-MDSC or tumor-MDSC (equal number of cells per each sample) was performed using mitochondrial isolation kit for mammalian cells (ThermoFisher) according to the manufacturer’s protocol. MtDNA from the cytosolic and mitochondrial fractions was isolated and purified using DNeasy blood and tissue kit (Qiagen). MtDNA was quantified by qPCR using primers listed in Table S1 and normalized to a nuclear DNA sequence (18s gene).
Mitochondrial DNA depletion
BM-MDSC were developed in the presence or absence of 150 ng/ml of ethidium bromide (Sigma Aldrich) for 4 days. On day 4, cells were collected and in vitro suppression assays were conducted as described above.
Immunofluorescence
Formalin fixed paraffin embedded TMA sections were stained using an automated OPAL-IHC system (PerkinElmer) in a BOND RX (Leica Biosystems). Briefly, slides were treated with the PerkinElmer blocking buffer for 10 min and incubated with the specific primary antibodies, followed by OPAL-HRP polymer and one OPAL fluorophore. Individual antibody complexes were stripped after each round of detection and DAPI applied as the last staining. Auto-fluorescence slides (negative control) included primary and secondary antibodies, omitting the OPAL fluorophores. Slides were imaged with a Vectra®3 Automated Quantitative Pathology Imaging System. Multi-layer TIFF images were exported from InForm (PerkinElmer) into HALO (Indica Labs) for quantitative image analysis. Each fluorophore was assigned to a dye color and positivity thresholds determined visually per marker based on nuclear or cytoplasmic staining patterns, and by intensity thresholds normalized for exposure (counts/2bit depth × exposure time × gain × binning area). Cell segmentation results from each core were analyzed using FCS Express 6 Image Cytometry (De Novo software).
Hematoxylin and Eosin (H&E) stained slides were scanned in an Aperio AT2 whole slide scanner (Leica Biosystems Inc.) equipped with a 20× 0.7NA objective lens. Images were created at 0.5 micron per pixel resolution and imported into Definiens Tissue Studio software v4.7 (Definiens AG) for analysis. Islets were found using a semi-automated segmentation process. First an automatic segmentation was applied to the image to create contour lines around objects within the image. Objects that contained islets were classified as such and remaining objects were classified as non-islet tissue. Next, adjacent objects of the same classification were merged together to clean up the segmentation. Segmented images were analyzed to provide the total area of islets. Immunofluorescence for mouse insulin in pancreas tissue sections was performed as we described (Sultan et al., 2017). Labeled samples were scanned with a Zeiss Imager Z2 Upright FL microscope with a 10x objective lens using the tile scan function. Images were created at a 0.65 micron per pixel resolution and imported into the Definiens Tissue Studio software v4.7 (Definiens AG) for islet detection and analysis. Islets were detected using a semi-automated segmentation process. First, an automatic segmentation was applied to the image to detect the tissue. Then, the software was trained to detect islet vs. non-islet tissue within the image. In order to remove false detected islets caused by background staining, detected islets larger than 150,000 square microns and less than 600 square microns were reclassified to non-islet tissue. Segmented images were then analyzed to provide the fluorescent intensity of islets and total tissue area.
Electron Microscopy
Tumor-MDSC were fixed in 4% paraformaldehyde, 2% glutaraldehyde in 0.1M sodium cacodylate (NaCac) buffer, pH 7.4, post-fixed in 2% osmium tetroxide in NaCac, stained in block with 2% uranyl acetate, dehydrated with a graded ethanol series, and embedded in Epon-Araldite resin. Thin sections were cut with a diamond knife on a Leica EM UC6 ultramicrotome (Leica Microsystems, Inc, Bannockburn, IL), collected on copper grids and stained with uranyl acetate and lead citrate. Tissue was observed in a JEM 1230 transmission electron microscope (JEOL USA Inc., Peabody, MA) at 110 kV and imaged with an UltraScan 4000 CCD camera & First Light Digital Camera Controller (Gatan Inc., Pleasanton, CA). Quantitative analysis was performed with ImageJ. (NIH, LOCI, University of Wisconsin-Madison)
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 7 (San Diego, CA). For two groups, means were compared by two-tailed unpaired Student’s t-test. For multiple groups and tumor growth studies, analysis was done by ANOVA with Bonferroni correction for multiple comparisons. P value of <0.05 was considered statistically significant. Specific statistical test results are indicated in each figure: *, p<0.05; **, p<0.01 ***, p<0.001.
Supplementary Material
Highlights:
Tumor-infiltrating MDSC demonstrate increased activation of PERK signaling
PERK ablation in MDSC elicits anti-tumor T cells and synergizes with immunotherapy
Deletion of PERK reprograms tumor-MDSC functionality by blunting NRF2 signaling
Reduced-NRF2 primes STING-to-Type I IFN axis in PERK-null MDSC via cytosolic-mtDNA
Acknowledgments
We thank the Flow Cytometry Core; the Tissue Imaging Core; and the Analytic Microscopy Core, all from Moffitt Cancer Center and partially funded through CCSG-CA076292. Also, authors thank J. Francis from the Lung Data Service at Moffitt.
Financial support: This study was supported in part by the National Institutes of Health (NIH) grants: R01CA184185 and R01CA233512 to PCR; R01CA103320 and R01CA211229 to DHM; and R01CA157664, R01CA124515, and R01CA178687 to JRCG. JRCR was supported by the Department of Defense (DOD) Ovarian Cancer Academy grant W81XWH-16-1-0438, the Mark Foundation ASPIRE Award, the Pershing Square Sohn Cancer Research Alliance, and the Stand Up to Cancer Phillip A. Sharp Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of conflict of interest
The authors declare no competing interests.
References
- Ahn J, Son S, Oliveira SC, and Barber GN (2017). STING-Dependent Signaling Underlies IL-10 Controlled Inflammatory Colitis. Cell Rep 21, 3873–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, and Barber GN (2014). Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 5, 5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. (2013). Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002. [DOI] [PubMed] [Google Scholar]
- Bettigole SE, and Glimcher LH (2015). Endoplasmic reticulum stress in immunity. Annu Rev Immunol 33, 107–138. [DOI] [PubMed] [Google Scholar]
- Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF, and Glimcher LH (2015). The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol 16, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, HuangFu WC, Dong G, Qian J, Baker DP, Karar J, Koumenis C, Diehl JA, and Fuchs SY (2013). Anti-tumorigenic effects of Type 1 interferon are subdued by integrated stress responses. Oncogene 32, 4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Trillo-Tinoco J, Sierra RA, Anadon C, Dai W, Mohamed E, Cen L, Costich TL, Magliocco A, Marchion D, et al. (2019). ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat Commun 10, 1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll EC, Jin L, Mori A, Munoz-Wolf N, Oleszycka E, Moran HBT, Mansouri S, McEntee CP, Lambe E, Agger EM, et al. (2016). The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity 44, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TK, Lawrence DA, Lu M, Tan J, Harnoss JM, Marsters SA, Liu P, Sandoval W, Martin SE, and Ashkenazi A (2018). Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol Cell 71, 629–636 e625. [DOI] [PubMed] [Google Scholar]
- Chevet E, Hetz C, and Samali A (2015). Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 5, 586–597. [DOI] [PubMed] [Google Scholar]
- Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, et al. (2016). Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, El-Deiry WS, Winograd R, Vonderheide RH, English NR, et al. (2014). ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J.Clin.Invest 124, 2626–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, and Gabrilovich DI (2009). Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J.Immunol 182, 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, et al. (2015). ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, and Diehl JA (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 279, 20108–20117. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, and Diehl JA (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23, 7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, Chen X, Sun JC, and Glimcher LH (2019). The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol 20, 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V (2012). Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambrock A, Loffler-Walz C, and Quast U (2002). Glibenclamide binding to sulphonylurea receptor subtypes: dependence on adenine nucleotides. Br J Pharmacol 136, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol.Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Hurst KE, Lawrence KA, Essman MT, Walton ZJ, Leddy LR, and Thaxton JE (2019). Endoplasmic Reticulum Stress Contributes to Mitochondrial Exhaustion of CD8(+) T Cells. Cancer Immunol Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakoshi NN, Pypaert M, and Glimcher LH (2007). The transcription factor XBP-1 is essential for the development and survival of dendritic cells. The Journal Of Experimental Medicine 204, 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Pulendran B, and Lambrecht BN (2014). Emerging functions of the unfolded protein response in immunity. Nat.Immunol 15, 910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, and Mellor AL (2016). STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res 76, 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P, and Ashkenazi A (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, Jiang S, Chang Q, Spring DJ, Sharma P, et al. (2017). Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 543, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas NL, and Diehl JA (2015). Molecular pathways: the PERKs and pitfalls of targeting the unfolded protein response in cancer. Clin Cancer Res 21, 675–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee A-H, and Glimcher LH (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature Immunology 11, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed E, Al-Khami AA, and Rodriguez PC (2018). The cellular metabolic landscape in the tumor milieu regulates the activity of myeloid infiltrates. Cell Mol Immunol 15, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, Laustsen A, Peri S, Hansen AL, Bonefeld L, et al. (2018). Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun 9, 3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, and Hotamisligil GS (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Puchalt A, Svoronos N, Rutkowski MR, Allegrezza MJ, Tesone AJ, Payne KK, Wickramasinghe J, Nguyen JM, O’Brien SW, Gumireddy K, et al. (2017). Follicle-Stimulating Hormone Receptor Is Expressed by Most Ovarian Cancer Subtypes and Is a Safe and Effective Immunotherapeutic Target. Clin Cancer Res 23, 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pytel D, Majsterek I, and Diehl JA (2016). Tumor progression and the different faces of the PERK kinase. Oncogene 35, 1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, Ochoa AC, Fletcher M, Velasco C, Wilk A, et al. (2014). Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int.J.Cancer 134, 2853–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, and Glimcher LH (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307. [DOI] [PubMed] [Google Scholar]
- Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, and Ochoa AC (2009). Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 69, 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Rivera D, Delvaeye T, Roelandt R, Nerinckx W, Augustyns K, Vandenabeele P, and Bertrand MJM (2017). When PERK inhibitors turn out to be new potent RIPK1 inhibitors: critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ 24, 1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, Brencicova E, Escovar-Fadul X, Nguyen JM, Cadungog MG, et al. (2015). Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 27, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, Shimoda M, Pacholczyk R, Shi H, Lee EJ, et al. (2018). Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c(+)CD103(+) Monocytic Antigen-Presenting Cells in Tumors. Immunity 48, 91–106.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, and Kensler TW (2007). NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 27, 7188–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra RA, Trillo-Tinoco J, Mohamed E, Yu L, Achyut BR, Arbab A, Bradford JW, Osborne BA, Miele L, and Rodriguez PC (2017). Anti-Jagged Immunotherapy Inhibits MDSCs and Overcomes Tumor-Induced Tolerance. Cancer Res 77, 5628–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Bhojnagarwala PS, O’Brien S, Moon EK, Garfall AL, Rao AS, Quatromoni JG, Stephen TL, Litzky L, Deshpande C, et al. (2016). Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 30, 120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Smith AL, Andrews KL, Beckmann H, Bellon SF, Beltran PJ, Booker S, Chen H, Chung YA, D’Angelo ND, Dao J, et al. (2015). Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase R-like endoplasmic reticulum kinase (PERK). J Med Chem 58, 1426–1441. [DOI] [PubMed] [Google Scholar]
- Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, et al. (2018). IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan H, Trillo-Tinoco J, Rodriguez P, and Celis E (2017). Effective antitumor peptide vaccines can induce severe autoimmune pathology. Oncotarget 8, 70317–70331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoronos N, Perales-Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, Nguyen JM, Curiel TJ, Cadungog MG, Singhal S, et al. (2017). Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov 7, 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, Ochoa AC, Cui Y, Del Valle L, and Rodriguez PC (2014). The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 41, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento C, Marigo I, Pievani A, Galleu A, Dolcetti L, Wang CY, Serafini M, Bronte V, and Dazzi F (2017). Bone marrow mesenchymal stromal cells induce nitric oxide synthase-dependent differentiation of CD11b(+) cells that expedite hematopoietic recovery. Haematologica 102, 818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D (2018). Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, Du X, Yang J, Li T, Wan Y, et al. (2018). Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity 48, 675–687 e677. [DOI] [PubMed] [Google Scholar]
- Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, et al. (2016). Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res 4, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wei L, Fan F, Ji S, Zhang S, Geng J, Hong L, Fan X, Chen Q, Tian J, et al. (2015). Integration of Hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat Commun 6, 6239. [DOI] [PubMed] [Google Scholar]
- Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li XD, Deng L, Chen ZJ, et al. (2017). Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363–373 e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wang HW, Bowman RL, and Joyce JA (2016). STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell Rep 16, 2914–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Zhao B, Gui J, Katlinski KV, Brice A, Gao Y, Li C, Kushner JA, Koumenis C, Diehl JA, and Fuchs SY (2015). Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc Natl Acad Sci U S A 112, 15420–15425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.